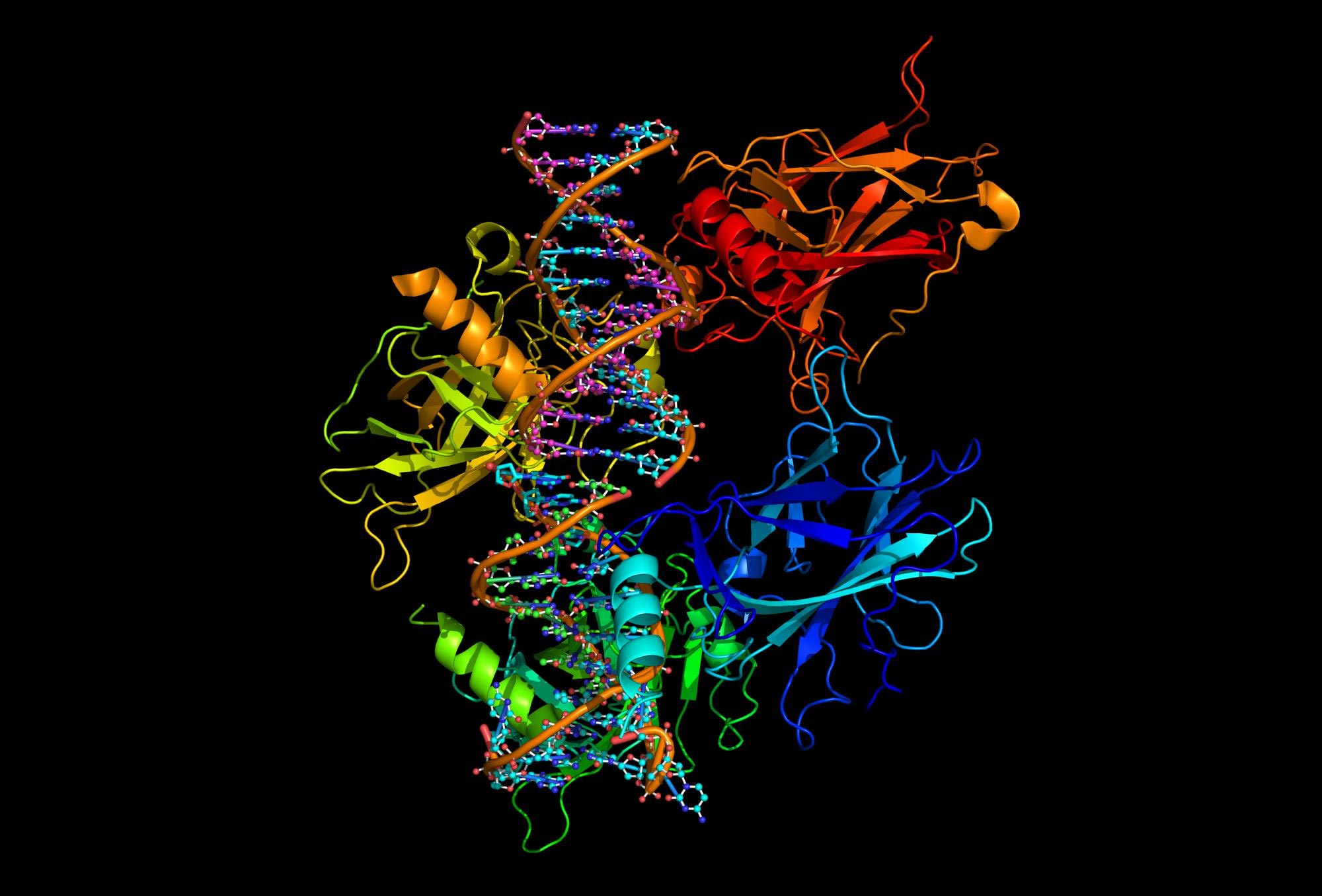
Harvard Medical School researchers have used evolution as a roadmap to determine the structure of 18 families of transmembrane proteins.
Scientists at Harvard Medical School developed algorithms that allowed them to use evolution as their guide to deduce the three-dimensional structure of 18 families of transmembrane proteins.
The molecules that drugmakers would most like to target are also among the hardest to study: Transmembrane proteins. These proteins connect our cells to their environment to sense, communicate and organize into tissue. A quarter of human proteins, transmembrane proteins constitute almost half of all drug targets. Knowing their shape is critical to drug design, yet solving the three-dimensional structure of these proteins is among biology’s hardest puzzles.
Using evolution as a guide, researchers at Harvard Medical School have deduced the shape of 18 families of transmembrane proteins. The findings, first published online May 10, will be reported in the June 22 issue of the journal Cell.
“The process of evolution and the power of sequencing technology have provided an unprecedented opportunity,” said senior author Debora Marks, an instructor in Systems Biology at Harvard Medical School. “These findings point to a fast, accurate method to understand the most interesting, difficult proteins and even predict their alternative conformations.”
Knowing the three-dimensional shape of protein molecules is key to understanding how they function, in both health and disease, and to designing drugs that target them. Normally the shape of protein molecules is determined by costly and complicated experiments, and for most proteins, including almost all transmembrane proteins, these experiments have not yet been done. Computing the shape of proteins from genetic information alone is possible in principle. But despite limited success with some smaller proteins, this challenge has remained essentially unsolved. The difficulty lies in the astronomically large number of possible protein shapes. Without any shortcuts, it would take a supercomputer many years to explore all possible shapes of even a small protein.
So Marks and colleagues developed algorithms — a set of shortcuts — that let them predict previously unknown 3-D transmembrane protein structures using information gleaned from conserved evolutionary patterns as raw material. While the 18 protein families had not been previously solved, they had tested previous predictions against known protein structures.
“One beauty of the evolutionary method is it doesn’t take expensive computers or computer scientists,” Marks said. “It’s very democratizing.”
The researchers caution that there are other limits, however: Experimental techniques for determining a protein’s structure, such as x-ray crystallography, are generally more accurate in atomic detail. (Marks is collaborating with experimental structural biologists to test approaches that combine her platform with these more traditional techniques.) And, the method works only when researchers have genetic data for large protein families. But advances in DNA sequencing have yielded a torrent of such data that is forecast to continue growing exponentially.
“As sequence technology continues to produce protein sequence information at an accelerating pace, a survey of an increasing fraction of the universe of protein structures and their interactions is within reach,” the researchers wrote.
Reference: “Three-Dimensional Structures of Membrane Proteins from Genomic Sequencing” by Thomas A. Hopf, Lucy J. Colwell, Robert Sheridan, Burkhard Rost, Chris Sander and Debora S. Marks, 10 May 2012, Cell.
DOI: 10.1016/j.cell.2012.04.012






Be the first to comment on "Evolution Helps Deduce the Shape of 18 Families of Transmembrane Proteins"