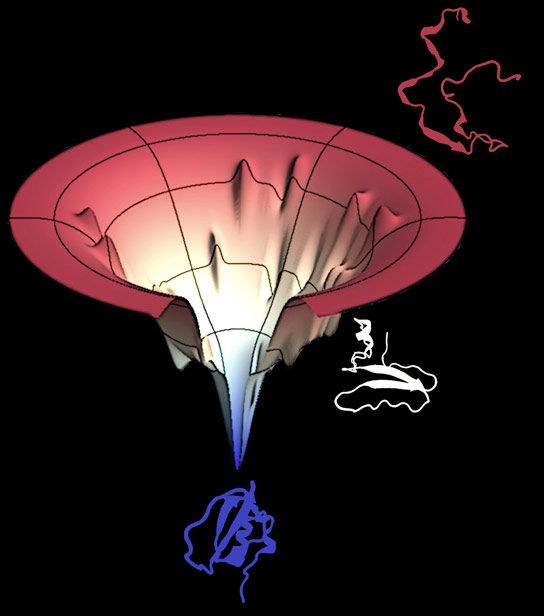
A protein folding funnel, the product of algorithms created by Rice’s Center for Theoretical Biological Physics, is a graphic representation of the energy landscape a protein navigates as it goes from its initial floppy state to a folded, functional unit. The funnel shows smooth slopes as well as outcroppings where parts of a protein may pause while others catch up, and also traps that could cause a protein to misfold. New Rice research shows how the interplay between evolution and physics developed the skills necessary to conserve useful proteins. Credit: Center for Theoretical Biological Physics
A team of biophysicists at Rice University used computer models to show that the energy landscape involved in the long-term evolution of proteins is essentially the same as that involved in the folding process that takes as little as microseconds.
Nature’s artistic and engineering skills are evident in proteins, life’s robust molecular machines. Scientists at Rice University have now employed their unique theories to show how the interplay between evolution and physics developed these skills.
A Rice team led by biophysicists Peter Wolynes and José Onuchic used computer models to show that the energy landscapes that describe how nature selects viable protein sequences over evolutionary timescales employ essentially the same forces as those that allow proteins to fold in less than a second. For proteins, energy landscapes serve as maps that show the number of possible forms they may take as they fold.
The researchers calculated and compared the folding of natural proteins from front to back (based on genomic sequences that form over eons) and back to front (based on the structures of proteins that form in microseconds). The results offer a look at how nature selects useful, stable proteins.
In addition to showing how evolution works, their study aims to give scientists better ways to predict the structures of proteins, which is critical for understanding disease and for drug design.
The research reported in the Proceedings of the National Academy of Sciences shows that when both of the Rice team’s theoretical approaches — one evolutionary, the other physics-based — are applied to specific proteins, they lead to the same conclusions for what the researchers call the selection temperature that measures how much the energy landscape of proteins has guided evolution. In every case, the selection temperature is lower than the temperature at which proteins actually fold; this shows the importance of the landscape’s shape for evolution.
The low selection temperature indicates that as functional proteins evolve, they are constrained to have “funnel-shaped” energy landscapes, the scientists wrote.
Folding theories developed by Onuchic and Wolynes nearly two decades ago already suggested this connection between evolution and physics. Proteins that start as linear chains of amino acids programmed by genes fold into their three-dimensional native states in the blink of an eye because they have evolved to obey the principle of minimal frustration. According to this principle, the folding process is guided by interactions found in the final, stable form.
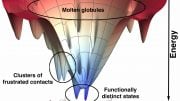
Wolynes used this fundamental law to conceptualize folding in a new way. The top of his folding funnel represents all of the possible ways a protein can fold. As individual stages of the protein come together, the number of possibilities decreases and the funnel narrows and eventually reaches its functional native state.
A funnel’s rugged landscape is different for every protein. It shows smooth slopes as well as outcroppings where parts of a protein may pause while others catch up, and also traps that could cause a protein to misfold.
“The funnel shows that the protein tries things that are mostly positive rather than wasting time with dead ends,” Wolynes said. “That turns out to resolve what was called Levinthal’s paradox.” The paradox said even a relatively short protein of 100 acids, or residues, that tries to fold in every possible way would take longer than the age of the universe to complete the process.
That may be true for random sequences, but clearly not for evolved proteins, or we wouldn’t be here. “A random sequence would go down a wrong path and have to undo it, go down another wrong path, and have to undo it,” said Wolynes, who in his original paper compared the process to a drunken golfer wandering aimlessly around a golf course. “There would be no overall guidance to the right solution.”
While Onuchic and Wolynes have been advancing their theories for decades, only recently has it become possible to test their implications for evolution using two very different approaches they developed on the shoulders of their previous work.
One of the algorithms they employ at Rice’s Center for Theoretical Biological Physics (CTBP) is called the Associative Memory, Water-Mediated, Structure and Energy Model (AWSEM). Researchers use AWSEM to reverse-engineer the folding of proteins whose structures have been captured by the century-old (but highly time-consuming) process of X-ray crystallography.
The other model, direct coupling analysis (DCA), takes the opposite path. It begins with the genetic roots of a sequence to build a map of how the resulting protein folds. Only with recent advances in gene sequencing has a sufficiently large and growing library of such information become available to test evolution quantitatively.
“Now we have enough data from both sides,” Wolynes said. “We can finally confirm that the folding physics we see in our structure models matches the funnels from the evolutionary models.”
The researchers chose eight protein families for which they had both genomic information (more than 4,500 sequences each) and at least one structural example to implement their two-track analysis. They used DCA to create a single statistical model for each family of genomic sequences.
The key is the selection temperature, which Onuchic explained is an abstract metric drawn from a protein’s actual folding (high) and glass transition (low) temperatures. “When proteins fold, they are searching a physical space, but when proteins evolve they move through a sequence space, where the search consists of changing the sequence of amino acids,” he said.
“If the selection temperature is too high in the sequence space, the search will give every possible sequence. But most of those wouldn’t fold right. The low selection temperature tells us how important folding has been for evolution.”
“If the selection temperature and the folding temperature were the same, it would tell us that proteins merely have to be thermodynamically stable,” Wolynes said. “But when the selection temperature is lower than the folding temperature, the landscape actually has to be funneled.”
“If proteins evolved to search for funnel-like sequences, the signature of this evolution will be seen projected on the sequences that we observe,” Onuchic said. The close match between the sequence data and energetic structure analyses clearly show such a signature, he said, “and the importance of that is enormous.”
“Basically, we now have two completely different sources of information, genomic and physical, that tell us how protein folding works,” he said. Knowing how evolution did it should make it much faster for people to design proteins “because we can make a change in sequence and test its effect on folding very quickly,” he said.
“Even if you don’t fully solve a specific design problem, you can narrow it down to where experiments become much more practical,” Onuchic said.
“Each of these methods has proved very useful and powerful when used in isolation, and we are just starting to learn what can be achieved when they are used together,” said Nicholas Schafer, a Rice postdoctoral researcher and co-author. “I’m excited to be participating in what I think will be an explosion of research and applications centered around these kinds of ideas and techniques.”
Faruck Morcos is the paper’s lead author and Ryan Cheng is a co-author. Both are postdoctoral researchers at Rice. Onuchic is Rice’s Harry C. and Olga K. Wiess Professor of Physics and Astronomy and co-director of the CTBP based at Rice’s BioScience Research Collaborative. Wolynes is the Bullard-Welch Foundation Professor of Science and a professor of chemistry and a senior scientist with CTBP.
The National Science Foundation, the National Institutes of Health, the CTBP, the Cancer Prevention and Research Institute of Texas and the D.R. Bullard-Welch Chair at Rice supported the research.
The researchers utilized the Data Analysis and Visualization Cyberinfrastructure supercomputer supported by the NSF and administered by Rice’s Ken Kennedy Institute for Information Technology.
Reference: “Coevolutionary information, protein folding landscapes, and the thermodynamics of natural selection” by Faruck Morcos, Nicholas P. Schafer, Ryan R. Cheng, José N. Onuchic and Peter G. Wolynes, 11 August 2014, PNAS.
DOI: 10.1073/pnas.1413575111






Be the first to comment on "Theorists Show How Energy Landscapes Dominate Both Evolution and Folding of Proteins"