
The flawed method has been used in hundreds of thousands of studies.
A new study reveals flaws in a common analytical method within population genetics.
According to recent research from Sweden’s Lund University, the most commonly used analytical method in population genetics is deeply flawed. This could have caused incorrect results and misconceptions regarding ethnicity and genetic relationships. The method has been used in hundreds of thousands of studies, influencing findings in medical genetics and even commercial ancestry tests. The findings were recently published in the journal Scientific Reports.
The pace at which scientific data can be gathered is increasing rapidly, resulting in huge and very complex databases, which has been nicknamed the “Big Data revolution.” Researchers employ statistical techniques to condense and simplify the data while maintaining the majority of the important information in order to make the data more manageable. PCA (principal component analysis) is perhaps the most widely used approach. Imagine PCA as an oven with flour, sugar, and eggs serving as the input data. The oven may always perform the same thing, but the ultimate result, a cake, is highly dependent on the ratios of the ingredients and how they are mixed.
“It is expected that this method will give correct results because it is so frequently used. But it is neither a guarantee of reliability nor produces statistically robust conclusions,” says Dr. Eran Elhaik, Associate Professor in molecular cell biology at Lund University.
According to Elhaik, the method contributed to the development of old beliefs about race and ethnicity. It plays a role in manufacturing historical tales of who and where people come from, not only by the scientific community but also by commercial ancestry companies. A well-known example is when a famous American politician used an ancestry test to back their ancestral claims prior to the 2020 presidential campaign. Another example is the misconception of Ashkenazic Jews as an isolated group or race driven by PCA results.
“This study demonstrates that those results were unreliable,” says Eran Elhaik.
PCA is used across many scientific fields, but Elhaik’s study focuses on its usage in population genetics, where the explosion in dataset sizes is particularly acute, which is driven by the reduced costs of DNA sequencing.
The field of paleogenomics, where we want to learn about ancient peoples and individuals such as Copper age Europeans, heavily relies on PCA. PCA is used to create a genetic map that positions the unknown sample alongside known reference samples. Thus far, the unknown samples have been assumed to be related to whichever reference population they overlap or lie closest to on the map.
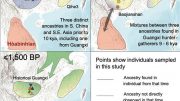
However, Elhaik discovered that the unknown sample could be made to lie close to virtually any reference population just by changing the numbers and types of the reference samples (see illustration), generating practically endless historical versions, all mathematically “correct,” but only one may be biologically correct.
In the study, Elhaik has examined the twelve most common population genetic applications of PCA. He has used both simulated and real genetic data to show just how flexible PCA results can be. According to Elhaik, this flexibility means that conclusions based on PCA cannot be trusted since any change to the reference or test samples will produce different results.
Between 32,000 and 216,000 scientific articles in genetics alone have employed PCA for exploring and visualizing similarities and differences between individuals and populations and based their conclusions on these results.
“I believe these results must be re-evaluated,” says Elhaik.
He hopes that the new study will develop a better approach to questioning results and thus help to make science more reliable. He spent a significant portion of the past decade pioneering such methods, like the Geographic Population Structure (GPS) for predicting biogeography from DNA and the Pairwise Matcher to improve case-control matches used in genetic tests and drug trials.
“Techniques that offer such flexibility encourage bad science and are particularly dangerous in a world where there is intense pressure to publish. If a researcher runs PCA several times, the temptation will always be to select the output that makes the best story”, adds Professor William Amos, from the Univesity of Cambridge, who was not involved in the study.
Reference: “Principal Component Analyses (PCA)-based findings in population genetic studies are highly biased and must be reevaluated” by Eran Elhaik, 29 August 2022, Scientific Reports.
DOI: 10.1038/s41598-022-14395-4







Elhaik is just a quack out to prove his discredited theories about Ashkenazi Jews being Khazars so he will stop at nothing to try to undermine the science that refutes him. The most dangerous scientist is the one with a political agenda when it comes to the results. That you are sharong his theories without due diligence on his background is morally repugnant.
I look forward to the response of Dr Elhaik’s peers to this radical claim that thousands of genetic studies are flawed due to PCA being a supposedly flawed methodof analysis. I would love to hear from Dr David Reich at Harvard especially.
Regrettably, Dr Elhaik is well known for his near obsession with Ashkenazi Jews and his contention that they are not descended from the historic Hebrew people of the Levant but instead are descended from the ancient Turkic tribe known as the Khazars who once lived in the area within modern day Ukraine. It will be interesting to see the reaction to Dr Elhaik’s latest paper from the scientific community.
I am curious if PCA will be replaced. I think it seems off as well, or maybe it’s results that are so back and forth. Ancestry said I am Ashkenazic. It said I’m Iberian, Moroccan, Sub-Saharan African, UAE, Saudi and Israeli, but removed those results after the first “update” came and I still have parts of my chromosomes that are basically not identified. I am very European-looking and lighter-complected. Overall, results across the internet are saying that I am those things that were removed from my initial test result. I have used the free portion of mytrueancestry and it shows a lot of ancient DNA remains and quite honestly from all over. That site and gematch both say I matched with Neanderthals and Denisovans, Iberians, lots of West Asians, Africans, all around the Mediterranean, Siberia, Greenland, etc. but not a lick of any of that is in my autosomal DNA results. I see that strides are being made with new developments in the genome project, in the studies and discoveries, but I’m perplexed how we can be traced back to a common ancestor 200,000 plus years ago, how I can match with Abusir el Meleq mummies, royalty from about 8 countries and so on and on. I’m wondering how or if or when such gaps could be closed in analysis and determination of reliable results. Learning about from these articles I’ve read on here, or at least this one, I thought it was being brought up that there are too many mistakes and most likely affecting hundreds of thousands of family tree projects. This is a lot of work for me. I have discovered I have a cognitive disorder, but I am hooked on a whole bunch of stuff surrounding DNA, ancient history, archaeology, etc. etc. When you read that you matched with remains near Gobleki Tepe and then lose that reference, it excites and then the feeling of being pied in the face. Any thoughts on an AI that can read data better? Anything?