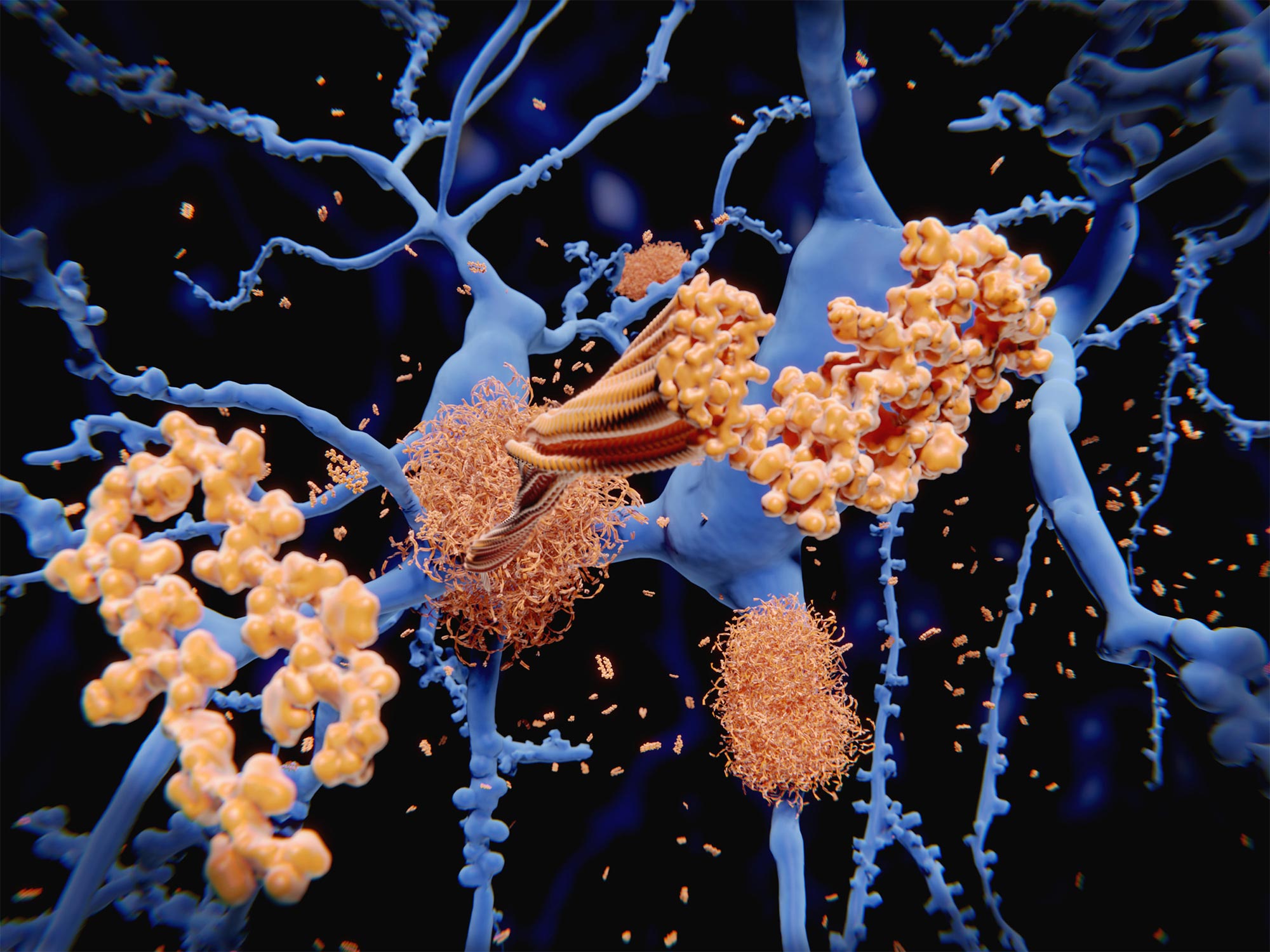
Amyloid protein (orange) forms clumps among neurons (blue). Amyloid in the brain is one of the proteins associated with Alzheimer’s disease.
In a major breakthrough, researchers have discovered how amyloid beta — the neurotoxin believed to be at the root of Alzheimer’s disease (AD) — forms in axons and related structures that connect neurons in the brain, where it causes the most damage. The findings could serve as a guidepost for developing new therapies to prevent the onset of this devastating neurological disease. The research, by scientists at Massachusetts General Hospital (MGH), was published in the journal Cell Reports.
Among his many contributions to research on AD, Rudolph Tanzi, PhD, vice chair of Neurology and co-director of the McCance Center for Brain Health at MGH, led a team in 1986 that discovered the first Alzheimer’s disease gene, known as APP, which provides instructions for making amyloid protein precursor (APP). When this protein is cut (or cleaved) by enzymes — first, beta secretase, followed by gamma secretase — the byproduct is amyloid beta (sometimes shortened to Abeta). Large deposits of amyloid beta are believed to cause neurological destruction that results in AD. Amyloid beta formed in the brain’s axons and nerve endings causes the worst damage in AD by impairing communication between nerve cells (or neurons) in the brain. Researchers around the world have worked intensely to find ways to block the formation of amyloid beta by preventing cleavage by beta secretase and gamma secretase. However, these approaches have been hampered by safety issues.
Despite years of research, a major mystery has remained. “We knew that Abeta is made in the axons of the brain’s nerve cells, but we didn’t know how,” says Tanzi. He and his colleagues probed the question by studying the brains of mice, as well as with a research tool known as Alzheimer’s in a dish, a three-dimensional cell culture model of the disease created in 2014 by Tanzi and a colleague, Doo Yeon Kim, PhD. Earlier, in 2013, several other MGH researchers, including neurobiologist Dora Kovacs, PhD (who is married to Tanzi), and Raja Bhattacharyya, PhD, a member of Tanzi’s lab, showed that a form of APP that has undergone a process called palmitoylation (palAPP) gives rise to amyloid beta. That study indicated that, within the neuron, palAPP is transported in a fatty vesicle (or sac) known as a lipid raft. But there are many forms of lipid rafts. “So the question was, Which lipid rafts? And which ones are most relevant to the neuronal processes making up the neural networks of the brain?” says Tanzi.
The new investigation revealed that palAPP is stabilized and prepared for cleavage by beta secretase in special lipid rafts within the neuron known as mitochondria-associated endoplasmic reticulum membranes (MAMs). “We showed for the first time not only that the MAM is where palAPP is processed by beta secretase to make Abeta, but that this happens exclusively in axons and neuronal processes where Abeta does most of its damage,” says Bhattacharyya, lead author of the Cell Reports paper. This role for MAMs was previously unknown, though earlier research indicated that they are increased in number and activity in the brains of people with Alzheimer’s disease.
Next, the MGH team wanted to learn what happens when MAM levels and activity were intentionally altered. They showed for the first time that preventing assembly of MAMs, either with gene therapy or a drug that blocked a key protein called the sigma-1 receptor (S1R), dramatically decreased beta secretase cleavage of palAPP in axons and lowered Abeta production. Conversely, a drug that activated S1R triggered an increase in beta secretase cleavage of palAPP and increased production of amyloid beta in axons.
“Our results suggest that the sigma-1 receptor might be a viable therapeutic target for reducing Abeta production, specifically in axons,” says Tanzi. The study also lends support for a strategy already under investigation by Tanzi and his team, which is developing an experimental treatment that inhibits the palmitoylation of APP, the process that produces palAPP. It’s also known that another class of drugs that Kovacs is studying for preventing formation of amyloid beta, called ACAT inhibitors, works directly in MAMs. In the future, these and other interventions that thwart production of this most dangerous pool of axonal amyloid beta could be used in concert with early detection (through blood or imaging tests) to stop or slow the progression of AD.
Reference: “Axonal generation of amyloid-β from palmitoylated APP in mitochondria-associated endoplasmic reticulum membranes” by Raja Bhattacharyya, Sophia E. Black, Madhura S. Lotlikar, Rebecca H. Fenn, Mehdi Jorfi, Dora M. Kovacs and Rudolph E. Tanzi, 18 May 2021, Cell Reports.
DOI: 10.1016/j.celrep.2021.109134
Tanzi directs the Genetics and Aging Research Unit and co-directs the Henry and Allison McCance Center for Brain Health at MGH and is the Joseph P. and Rose F. Kennedy Professor of Neurology at Harvard Medical School (HMS). Bhattacharyya is also an instructor in Neurology at HMS.
This study was funded by grants from the National Institutes of Health and the Cure Alzheimer’s Fund.






Helpful article. I found it odd that the author interrupted the scientific text to divulge the marital status of the team’s female researcher – unnecessary and unprofessional.
Mentioned her marital status?
Oh the humanity!
Very interesting and vital work. A well constructed article but why divulge the marital status of members of the scientific team?
Phenomenal discoveries. It’s very inspiring to know a Husband and Wife are on the same team following their dreams to help people together.
Does anyone Remember the movie Awakenings with Robin Williams, based on a true story. I wonder if that drug used was something that could be researched today that could have some impact on these type patients, to basically see if it helps “clean up or Clear out these lipid rafts or proteins that are causing this affect on the Brain ??? Just a Crazy thought when I read this article! ????
Just sounds like another money drug instead of ways to prevent. It seems that research is geared for profit.
Full disclosure: this neurobiologist is married and is therefore… what, exactly?
So true regarding the marriage. If only she properly took his surname, for what ever reason the author included that point, a simple Mrs. would have been sufficient. I’m only a simple person interested in the prevention of AD, but I seem to recall my wife of 39 years didn’t have any trouble taking on my name.
Can you put in laymen terms so more can understand please
Mentioned her marital status?
Oh the humanity!
Mom is in early stages of Alzheimer’s, we live in Florida, but is there any way, I can get her involved with the new studies for a cure? We love her so much.
I wonder if this disease is largely caused by the first world diet. It would be interesting to look at rural China, aborigines in Australia, and small tribes across the African continent to see if the disease is present and to what degree.
It’s great that we can look into the science, but to think we can just make a pill to solve the problem is quite naive. Drug industries will profit the greatest if the disease is “managed”. If the drug could actually cure the disease, the companies would lose their patients. I’m glad teams can fulfill the realization of their life’s work, but it won’t be to cure the disease, no matter how good intentioned the research teams are.
I am not a fan of some of the economic considerations involved in big Pharma and there are certainly some unethical practices in the world. However, to completely discount anything that comes in a pill is wrong. Are you aware that penicillin and its variants come in pill form and the countless lives saved from infection thanks to those pills? There are hundreds of diseases that are no longer a threat to mankind thanks to pills and injections. Have you ever taken an antibiotic or antiviral pill? I have and I was cured of disease and I’m grateful that the pill exists.
I am very interested in the progress of this study because I lost my grandmother and mother to Alzheimer’s. I also carry the Alzheimer’s gene and I’m looking at a 20-year countdown until I am at the age when they both suffered their fate. The last thing I need are a bunch of anti-vaxxers who don’t understand basic concepts to start slowing down research because they are upset that someone can earn a living at their very helpful profession.
Never knew anyone with this that wasn’t a multi tasker , with a lot of responsibility.
Followers usually don’t have this problems. ( Just observation in my 69 years )
Is that a snowflake by your name? I found the part where they said they couldn’t go down a certain road due to safety issues interesting
Bravo for letting it be known that females are just as brilliant as men doing such cutting edge research. I hope they do likewise when Blacks, Latinos, etc are involved.
Yes he mentioned the marital status of the female colleague but she is his wife.
Funny that someone would read about this amazing work and be offended at the mention that the researchers were married. Actually, funny isn’t the word that’s comes to mind….
I agree, Why mention the marital status of the 2. It really interrupted my focus on the whole. Being I’m lightly educated in the terminology and understanding of the humane body. Stay on point.
I really thought it was horrific and oh, so 19th century of you to mention a woman’s marital status in a scientific article. You did not mention her husband’s marital status. It is so disheartening. Will things ever change?
Great breakthrough… by another hopefully celebrated wife and husband team… I think that was the only reason to mention it… how advantageous to go to bed discussing your work and results with another passionate collaborative…✌
Hallelujah….praises to those dedicated to this💯‼️ Thank you!!😊
The scientific text was not interrupted. The fact that they are married was part of listing some lab personnel involved in the previous research. It does seem relevant that they are married,and just as it would if they were siblings or parent/child, makes for a better story. To single this out as worth commenting on, in light of the achievements of that team, shows how “outrage culture” can needlessly bleed even into scientific discussions.
Did the study leave out dietary components? Or did I skim over the science speak and miss it?
mentioning that two of the researchers are married triggered so many people? wow Karens really need a life.
How sad that the only thing some people zoomed in on was a marital status. And the one comment that they didn’t mention the HUSBAND’S marital status, as well? DUH, the phrase (who is married to Tanzi) discloses his marital status! People (generally snowflakes) who pick through an article to find anything to be offended about are obviously not concerned with the progress being made against this insidious disease & most likely haven’t had a loved one suffer & die from it. Get over yourselves & appreciate what these scientists have done. I’m not a fan of Big Pharma either, but would sell everything I own to pay for a treatment/cure for my 2 family members to have enjoyed longer lives without suffering.
Congratulations to all who contributed to this research…including the married couple!
To the commentor, “Independent Married Woman:
“Full disclosure: this neurobiologist is married and is therefore… what, exactly?”
Maybe you should lose the “Married” word in your comment name. What purpose does it serve??
And commentor Robert Belcher:
“Just sounds like another money drug instead of ways to prevent. It seems that research is geared for profit.”
Honestly, what would your suggestion be for a FREE solution to prevent Alzheimer’s, hmmm?
You triggered folks are so entertaining!
Great research and work in identifying viable targets for drug development. Making a safe drug won’t be easy since shutting down the enzyme responsible for cleavage is not an optimal solution. a-secretase or g- secretase might be better investigating. Blocking BACE1 has failed in recent studies. Boosting a-secretase ..look at that one!
Doesn’t matter in the least the mention of a marital status.
Thank you for your continued efforts over the years!!! This sounds like an amazing breakthrough. Please keep up your good work. To finally know what is causing this devastating disease is miraculous!
The REASON they mention the marital status is because they have different last names. It happens for many good reasons, and none of them have to do with your business. Get OVER yourself.
I don’t care about whom is married to whom. I want to know how this can affect my future. My mother is struggling with Alzheimers. It’s too late for her. I wonder what can be done to prevent this horrible disease.
Ya oxidative stress and lack of k2 and anything that speeds system steroids herbicides medication anything that cause oxidative stress causes cancer and many other diseases
Just here to point out that “Sid” was so irritated by ppl not appreciating the disclosure of marital status that they literally posted the same comment twice.🤣
“Oh the Humanity”
Interesting article, appears to give the history of how the team got to this point and genetic and molecular basis of what is involved. These neurobiologidts are looking for pathways and what causes. Still premature to determine if diet and lifestyle. Whether two people are married shows a human side. The fact is science is hard. Someone mentioned ethnicity and that us irrelevant. Maybe instead of focusing on gender and there only two genders or race we can elevate our discussion to points of finding a cure.
Still a solid generation 25 years before there is any measurable cure. The complexity of the disease highly variable. A common thread in dementia and Alzheimers is diabetes.
This is news or theory? The husband and wife mention is disturbing and shockingly amature. I don’t see anything in this development but plack diagnosis
Mentioned the fact that the two doctors researching this were married? OMG ! Don’t you know.. scientists don’t get married and the have babies by test tube. People will complain about anything.🤣
Read the following regarding the real truth about sigma 1 receptor agonists for the treatment of Alzheimer’s disease, Parkinson’s disease, Rett syndrome and various other CNS conditions and diseases.
https://e1cd7807-443e-425e-9433-41548681800c.filesusr.com/ugd/73f698_021c7a1cb7a24d78b02dac05e7262ef7.pdf
Watch the following short news video:
https://www.youtube.com/watch?v=8orzf4UXNLA
Good luck and GOD bless,
After watching my grandma and now my mother suffer from this God Awful less than Humane disease.. I’m glad there is something promising..I would take chicken boo boo at this point not to have it…
he Forgotten Life of Einstein’s First Wife
She was a physicist, too—and there is evidence that she contributed significantly to his groundbreaking science
https://blogs.scientificamerican.com/guest-blog/the-forgotten-life-of-einsteins-first-wife/
Both men and women develop scientific discoveries.
Good luck and GOD bless,
IAM 69 YRS OLD MY MON STARTED WITH THE ALZHEIMER at 80 she die @93 I supose me and sis will have I only have a few yrs left in english did you find anything in your studies that cN prevent it from happening I have for past 7 yrs chrone disease have a permanent bag for inflation I would like to know what are my chances and if is a bio med like the one iam taking every 8 weeks for it.thayou waiting for a respond
Disclosing that the scientists are married is there to reveal potential bias. Had her marital status been to someone completely unrelated, then yes unnecessary.
Just going to say it’s pretty sad so many are offended by the fact that the author noted that 2 of the researchers are married. Imagine the horror those poor people commenting are going through to discover that a husband and wife can work as a team on anything. Even more shocking to them I guess is the fact that it’s to end suffering on victims of a disease that ravages millions of families and their loved ones. I wonder if maybe they can come up with a pill to help the stupid that are worried marriage might become a “thing” again.
Idiots.
Focus should be upon scientific research. When I read these articles, I try to truly understand the information.
I feel that disclosure of a marriage between 2 people was for legal protection.
If you cannot contribute knowledge as to how Thi research might be moved forward for treatment, go whine about something controversial in our new found culture wars.
I have a father in law who is getting worse in his battle with this disease. Subjects should be studied in all phases/age groups. Anyone want to help in this faction? I am an ideal candidate to be studied in the field of neurology as well. Please contact me. Maybe studies of “Woody” and my own issues as a long term prescription user for Prozac, Clonazepam, Buspirone, and Trazodone could help someone else. Look up my physical address, call me. I want to help others.
770.595.7752. I rarely use my email.
Look at Anavex Life Sciences (AVXL) that is developing a drug molecule Blarcarmesine which activates the Sigma 1 Receptor. They are now in phase 2/3 clinical trials in the UK, Germany and Australia and patients who have completed phase 1 and 2 have continued taking the drug (OLE). No safety issues in either phase 1 or 2 and data should be available same time next year, after a 48 week trial duration.
Interestingly, the study showed that “activation” of the sigma-1 receptor protein resulted in increased production of amyloid beta. Looks like that particular protein in neurons is a culprit in Alzheimer’s disease.
However, another sigma-1 receptor activator, Anavex Life Science’s blarcamesine (Anavex 2-73), does just the opposite. Its activation of the protein causes the protein to take on or resume modulation or facilitation of a number of homeostatic processes in the neuron. With those, the neuron functions normally, Alzheimer’s symptoms abate (or are prevented).
Scrutinize the schematics on Anavex sigma-1 receptor activation here:
https://www.anavex.com/sigmaceptor-explained
ANAVEX®3-71, which targets sigma-1 and muscarinic receptors, is a promising clinical stage drug candidate demonstrating disease-modifying activity against the major hallmarks of Alzheimer’s disease in transgenic (3xTg-AD) mice, including cognitive deficits, amyloid, and tau pathologies. In preclinical trials, ANAVEX®3-71 has shown beneficial effects on mitochondrial dysfunction and neuroinflammation.
Another article that proves more research needs to be done. Dozens of other researchers have shown that amyloid-beta does not cause AD. It may be associated with it, but doesn’t play as large a role as once thought