
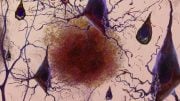
MIT researchers have demonstrated a new way to reverse memory loss by blocking an enzyme known as HDAC2. Credit: Jose-Luis Olivares/MIT
Research from MIT suggests a new approach to developing treatments for Alzheimer’s disease, revealing a new way to reverse memory loss by blocking an enzyme known as HDAC2.
In the brains of Alzheimer’s patients, many of the genes required to form new memories are shut down by a genetic blockade, contributing to the cognitive decline seen in those patients.
MIT researchers have now shown that they can reverse that memory loss in mice by interfering with the enzyme that forms the blockade. The enzyme, known as HDAC2, turns genes off by condensing them so tightly that they can’t be expressed.
For several years, scientists and pharmaceutical companies have been trying to develop drugs that block this enzyme, but most of these drugs also block other members of the HDAC family, which can lead to toxic side effects. The MIT team has now found a way to precisely target HDAC2, by blocking its interaction with a binding partner called Sp3.
“This is exciting because for the first time we have found a specific mechanism by which HDAC2 regulates synaptic gene expression,” says Li-Huei Tsai, director of MIT’s Picower Institute for Learning and Memory and the study’s senior author.
Blocking that mechanism could offer a new way to treat memory loss in Alzheimer’s patients. In this study, the researchers used a large protein fragment to interfere with HDAC-2, but they plan to seek smaller molecules that would be easier to deploy as drugs.
Picower Institute postdocs Hidekuni Yamakawa, Jemmie Cheng, and Jay Penney are the lead authors of the study, which appears in the August 8 edition of Cell Reports.
Memorable interactions
In 2007, Tsai first discovered that blocking HDAC activity could reverse memory loss in mice. There are several classes of HDACs, and their primary function is to modify histones — proteins around which DNA is spooled, forming a structure called chromatin. These modifications condense chromatin, making genes in that stretch of DNA less likely to be expressed.
Human cells have about a dozen forms of HDAC, and Tsai later found that HDAC2 is responsible for the blockade of memory-linked genes. She also discovered that HDAC2 is elevated in human Alzheimer’s patients and in several mouse models of the disease.
“We think that HDAC2 serves as a master regulator of memory gene expression, and during Alzheimer’s disease it’s elevated so it causes an epigenetic blockade of the expression of those memory genes,” she says. “If we can remove the blockade by inhibiting HDAC2 activity or reducing HDAC2 levels, then we can remove the blockade and restore expression of all these genes necessary for learning and memory.”
Most of the existing HDAC inhibitors that block HDAC2 also affect HDAC-1, which can have toxic side effects because HDAC1 is necessary for cell proliferation, especially in the production of white and red blood cells.
To find a way to more specifically target HDAC2, Tsai set out to identify proteins that help the enzyme bind to genes required for memory formation. First, she analyzed gene expression data from postmortem brain samples taken from people who did not have Alzheimer’s disease, including 28 brains with high HDAC-2 levels and 35 with low levels. This search yielded more than 2,000 genes whose levels closely matched HDAC2 levels, suggesting that those genes might work in tandem with HDAC2.
Based on what they already knew about these genes’ functions and how they physically interact with HDAC2, the researchers then picked out three of those genes for further testing. Those tests revealed that a gene called Sp3 is necessary to recruit HDAC2 to chromatin to enact its blockade of memory-linked genes.
The researchers also examined gene expression data from postmortem brains of Alzheimer’s patients and found a nearly perfect correlation between levels of HDAC2 and sp3.
Specific targets
The researchers then explored what would happen if they lowered Sp3 levels in a mouse model of Alzheimer’s disease. In these mice, the same type in which they previously studied the effects of blocking HDAC2, they found that deactivating Sp3 also restored the mice’s ability to form long-term memories.
The researchers used a type of short RNA strand to perform the genetic “knockdowns” in these experiments, but for this approach to be useful for potentially restoring memory function in human patients, scientists would likely need to develop a drug in the form of a small protein or chemical compound.
To that end, the researchers identified the section of the HDAC2 protein that binds to Sp3. When they engineered neurons to overproduce that HDAC2 fragment, the fragment sopped up most of the available Sp3, blocking it from binding HDAC2 and releasing the blockade of memory-linked genes. Furthermore, the fragment did not interfere with cell proliferation, suggesting that this more targeted approach would not have the adverse side effects of more general HDAC inhibitors.
“This therapeutic approach is specific to the action of HDAC2 and does not affect other HDACs, such as the close homologue HDAC1. The data raise hopes that therapeutic strategies targeting Sp3 or the interaction of Sp3 with HDAC2 may overcome the issue of lacking specificity of HDAC2 inhibitors,” says Andre Fischer, a professor at the German Center for Neurodegenerative Diseases, who was not involved in the study.
The protein fragment that the researchers used to block the interaction in this study has about 90 amino acids, which would likely be too large to use as a drug, so the researchers hope to either identify a smaller segment that would still be effective, or find a chemical compound that would also disrupt the Sp3-HDAC2 interaction.
Tsai also hopes to further investigate some of the other genes that were found to correlate with HDAC2, in hopes of identifying other drug targets. She also plans to explore whether this approach could be useful in treating other disorders that involve elevated levels of HDAC2, such as posttraumatic stress disorder.
Reference: “The Transcription Factor Sp3 Cooperates with HDAC2 to Regulate Synaptic Function and Plasticity in Neurons” by Hidekuni Yamakawa, Jemmie Cheng, Jay Penney, Fan Gao, Richard Rueda, Jun Wang, Satoko Yamakawa, Oleg Kritskiy, Elizabeta Gjoneska and Li-Huei Tsai, 8 August 2018, Cell Reports.
DOI: 10.1016/j.celrep.2017.07.044
The research was funded by the Robert and Renee Belfer Family Foundation.




I’m not sure why this is news as an association between memory and HDAC2 has been known since 2007. Moreover, HDAC2 (as with all HDACs) is a master regulator, so it’s not exactly clear how you can perturb it without having a ton of other off target (side) effects. Thus the impact of discovering that HDAC2 is involved in memory is nearly equivalent to discovering that neurons are involved in memory loss — almost to general to be useful..
The novelty lies in the discovery of SP3 being a necessary partner of HDAC2 inhibition, and thus the suppression of that, as done here by binding with non-active HDAC2 fragments, would render this process very specific to HDAC2 only.