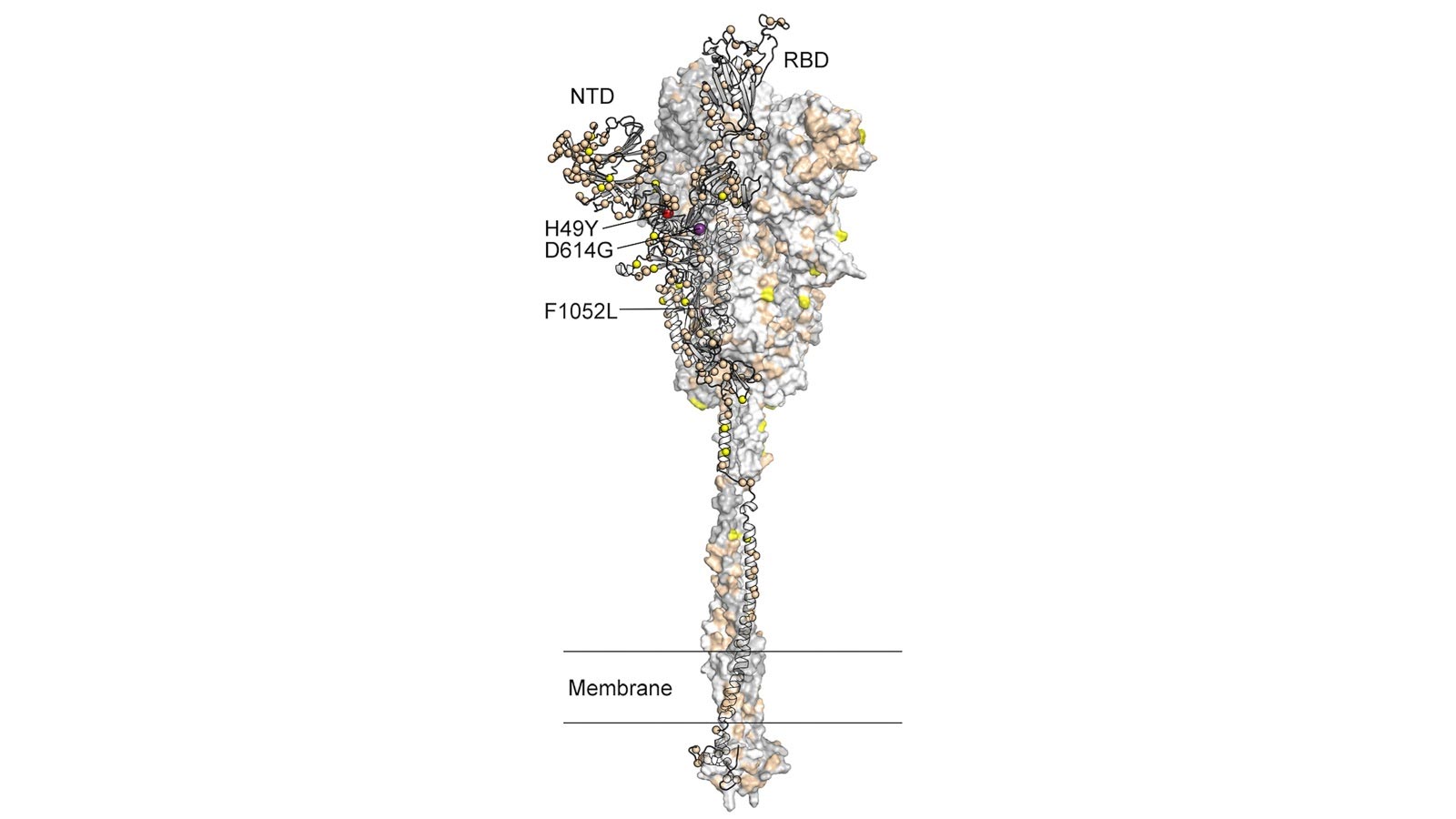
Amino Acid Substitutions on SARS-CoV-2 Spike Protein
Argonne computational resources supported the largest comprehensive analysis of COVID-19 genome sequences in the U.S. and helped corroborate growing evidence of a protein mutation.
Before COVID-19 first entered the United States in March, Houston Methodist Hospital had already begun preparations to test for and sequence the virus on a large scale, given the news coming out of Wuhan, China.
Between March 5, when the first case turned up in metropolitan Houston, and July 7, physicians/researchers at Houston Methodist sequenced the genome of over 5,085 strains of the virus. These accounted for nearly 10 percent of the COVID-19 cases that came through the 2,400-bed Houston Methodist health system, during two distinct waves that occurred in that time frame.
“99 percent isn’t 100 percent. If there is a mutation that accounts for just one percent of the population, and you suppress or eradicate the majority, you can drive up some trait of that one percent, whether it’s virulence or transmissibility, and then it’s a different ballgame.” — James Davis, Argonne staff scientist
Collaborators from the University of Texas at Austin, Weill Cornell Medical College, the University of Chicago and the U.S. Department of Energy’s (DOE) Argonne National Laboratory worked together to analyze the data and try to correlate patient outcomes with viral traits.
“This is the largest viral sequence analysis in the U.S. right now and it’s one of the most comprehensive, continual snapshots of sequences that dates to the beginning of the outbreak,” said James Davis, a staff scientist in Argonne’s Data Science and Learning division. “It also provides a much clearer picture of how the strains are evolving.”
During the course of the research, the group helped solidify mounting observations and concerns internationally that a mutation in the virus’s spike protein had become dominant, driving COVID-19’s transmissibility rates as witnessed by the second wave that surged through Houston around mid-May.
A paper describing their methods and results was published in the journal mBio on October 30 ,2020.
That mutation in the spike — responsible for infiltrating the human immune system and the current target of vaccine research — was in an amino acid called Gly614 and was the result of one protein, aspartic acid, mutating into another, glycine.
During the earliest parts of the pandemic, March through April, Gly614 was just one variant among many others. But during the second wave in May, Davis recalls, all of the cases they were sequencing at Houston Methodist showed that Gly614 had proliferated to the point of becoming the dominant amino acid in the spike protein.
In fact, it was found in over 99 percent of the sequenced variants.
“The SARS-CoV-2 virus is remarkably conserved, so whenever you see changes like this it’s more remarkable because you don’t tend to see that many mutations,” he said. “I’m not sure if it makes the virus more virulent or easier to transmit, but the study does show some data suggesting that patients with the Gly614 mutation have a larger viral load, though they aren’t necessarily sicker.”
Coincident with the Gly614 takeover in the second wave, patients tended to be younger, showed less severe symptoms, were more likely to be Hispanic/Latino, and lived in areas of lower median incomes. Still, the reasons were unclear and they’d hoped that Argonne’s computational resources would open a door on the causes.
A working relationship already in place, Houston Methodist approached Argonne for help with genome sequence analyses of the 5,000-plus COVID strains, as well as phylogenetic analyses, which look at changes in an organism or a specific feature over time.
Through its Bioinformatics Resource Center Project, supported by the National Institute of Allergy and Infectious Diseases, Argonne provides computational and technical resources to collaborators conducting large biological-based data projects. In this case, it managed the sequencing components, and included quality control, genome alignments, and the building of phylogenetic trees.
“For a virus, it has a pretty big genome,” noted Davis, “so, the process became computationally expensive.” But with its arsenal of computers, both large and super, Argonne was prepared to handle the influx of data.
Another aspect of that work involved the artificial intelligence technique called machine learning. While the focus of machine learning among many research institutions, including Argonne, has focused on determining how drugs might interact with COVID-19, Marcus Nguyen hoped to predict whether the sequence of the virus could ultimately predict patient outcome or patient demographics.
Nguyen, a research specialist with a joint appointment at Argonne and the University of Chicago, looked at correlations between the genome sequences and patient information.
The process trained a machine learning algorithm on the genomic sequences from Houston Methodist, as well as past patient outcomes — length of hospital stay, need for mechanical ventilation, mortality — to potentially determine those outcomes.
“Unfortunately, we didn’t get the results we were hoping for,” said Nguyen. “Although there have been a few correlations found in different pieces of patient metadata, I don’t think there is anything in the genome that is indicative of patient outcome, so there has to be something else going on.”
While the group did observe other variations in the spike, research continues on Gly614 to understand its dynamics and determine what role, if any, it might play in treatment therapies.
“It should be helpful in vaccine development because it shows so many different sequences and so many variants that you get a pretty good picture of what to look for in terms of what variants are dominant in the population,” said Davis.
Though the virus has remained somewhat stable, to date, this study — and natural history — have shown that it only takes one mutation to create a powerful impact on life, both large and diminutive.
“99 percent isn’t 100 percent,” Davis pointed out. “If there is a mutation that accounts for just one percent of the population, and you suppress or eradicate the majority, you can drive up some trait of that one percent, whether it’s virulence or transmissibility, and then it’s a different ballgame.”
This research appears in the article “Molecular Architecture of Early Dissemination and Massive Second Wave of the SARS-CoV-2 Virus in a Major Metropolitan Area,” in mBio, October 30, 2020. Maulik Shukla, of both Argonne and the University of Chicago, was among the co-authors.
Read Coronavirus Genetic Mutation May Have Made COVID-19 More Contagious for more on this research.
Reference: “Molecular Architecture of Early Dissemination and Massive Second Wave of the SARS-CoV-2 Virus in a Major Metropolitan Area” by S. Wesley Long, Randall J. Olsen, Paul A. Christensen, David W. Bernard, James J. Davis, Maulik Shukla, Marcus Nguyen, Matthew Ojeda Saavedra, Prasanti Yerramilli, Layne Pruitt, Sishir Subedi, Hung-Che Kuo, Heather Hendrickson, Ghazaleh Eskandari, Hoang A. T. Nguyen, J. Hunter Long, Muthiah Kumaraswami, Jule Goike, Daniel Boutz, Jimmy Gollihar, Jason S. McLellan, Chia-Wei Chou, Kamyab Javanmardi, Ilya J. Finkelstein and James M. Musser, 30 October 2020, mBio.
DOI: 10.1128/mBio.02707-20
Research funding was provided by the National Institutes of Health (NIH) National Institute of Allergy and Infectious Diseases (NIAID) Bacterial and Viral Bioinformatics resource center.






How do I unsubscribe?
The Covid 19 epidemic, like Christianity, is founded on superstition and fear.
Actually, Christianity is based upon love.
Hahahahahahahahaha!!! Another scientific talk ran over peoples political OPINION! OPINIONS are like your lower orifice, you all have one and they All stink. Keep your opinion where they belong. This is science. And yet noted many incorrect statements. All living entities on Earth used DNA in a form of nucleus or nucleolus. However, virus have single strand or double stranded DNA or RNA.
Hahahaha “you all have one and they all stink”
Butt of course mine doesn’t.
And science is not the “study of what is” no no, it’s what I want to call it because I don’t understand the study of creation is science.
Just like geology is a science , theology is a science. Proctology is a science but mine doesn’t stink. Hahahaha.
“…was in an amino acid called Gly614 and was the result of one protein, aspartic acid, mutating into another, glycine.”
Think you have that a tad backwards. Glycine and aspartic acid are amino acids which form proteins. Gly614 refers to the mutation (polymorphism) wherein aspartic acid has been replaced by glycine, at the 614th amino acid in the sequence for the spike protein.
Covid is the 2nd biggest hoax right next to man made climate change. Created by China and with the help of Democrats this weak version of influenza was blow up to get trump out of white house. It worked as dems pushed mail in voting and created massive voter fraud. America is now going to be ran by evil socialists who want total controle. America is finished.
This is the end of man kind an it not because of just one man that took on a country when he didn’t have to. Didn’t want our money he just want to help, an just he know something we don’t know, with the first day he took office the wall will be built, an as we know now we need it, I really think the democrats read this election with mail in ballots because the older your ballot went in by that date of election and then count and by that Trump will be our president, that’s how we are the Great Falls the bed risers in America we had eight years of democrat ran Country in the ground. What makes you think it ain’t happening again, now the reason we have this COVID-19 daily disease mutation anything we treat it with because God put it here mother nature she keeps on lashing out on us 300,000 years is earth been here we destroy it this time, So I say goodbye to everybody what we created can’t be destroyed this time
Biden would have nothing to run on if Covid 19 scam didn’t show up! Talk about last second hail Mary! You could tell who was for Biden just by who was wearing masks! Show your hands if you want Joe… Every mask wearing TDS Trump hater raises their hands!
You know??? The thing? Come on man, you on drugs? Now we brace ourselves for the impact of Harris who has been soundly rejected by America with maybe 2 percent of the vote with her failed attempt at the DNC nomination now running the country she can open the boarders abolish the police take your guns (she said so herself) and continuing to murdering countless millions of unborn babies! Next she is giving social security to illegal aliens with free health care! Pelosi will have her evil plans with the ingnorant Shummer and Shiftless Shift promoting her every wicked socialist agenda!
Tell that to the 225,000 families whos holidays will look much sadder this year. This is why it has been so much worse than it could have been here. The entire world was not in on a conspiracy to get rid of Trump. Let’s stick to the science shall we.
To think that I served two tours in Vietnam to keep from having to fight the communists on the beaches of Santa Monica. Boy, that worked out well.
“Before COVID-19 first entered the United States in March, ” Really – in March? Really?
53 years and the only president that knew what it “really takes” to run this country Like a profitable business. But nor to the point he understood what “Anerica” means. He is the ONLY president to ever enact, execute, accomplish, stand up to an “DO” things in the best interest of the American dream. He want in anyone’s pocket, didn’t owe everyone a favor for getting him elected.he didn’t make policy based in his own egotism or enact laws to further a political parties agenda. He was a renegade for those who remember what the American Dream used to look like.
I feel the same way Trump was the best thing for this country and he knew how to run a business and that’s why we needed him with COVID-19 it to me he still defeated it and when people say blame on him they’re just a bunch of mother****ing dumb***es
All you Trumper idiots are showing your ignorance. Finally there is some sanity and I can wake up every day and not have to worry about what new personal trivial president grievance the country is trying to grapple with. Or what new non sensical suite of 4 year old – level of sophistication set of lies and accusations come out to help cover up personal responsibility.
The fact is, the Corona Virus pandemic is the first real Crisis the president had to deal with that he hadn’t manufactured himself.
He didn’t create the crisis. But he did nothing to stop it. Replace it with any other thing and the results would be the same.
Instead of Covid, imagine if it was a fire, drought, rats,100K daily massive personal attacks by hackers on random people.
His response was to do nothing. Worse than that, he undermined people who actually know what they are talking about and crippled our ability to resolve it ourselves.
Every other president has had to deal with multiple real problems that came about unexpectedly. For most of Trump’s presidency, he’s had nothing real to deal with. He was handed an economy that was smoothly chugging along. Everything was in place. A real problem comes about and its treated like a campaign issue instead of an actual thing that should be addressed. Thank god we didn’t have any more crises because this one crisis shows that he has no ability to lead anything to victory.
No wonder the country said “You’re Fired!” and kicked him to the curb. The scary part Is that there are so many people willing to let it get to this level. Sure, he’s still in office until early next year. But he doesn’t have a campaign to worry about so he can focus on doing what he does best … lawsuits, talking nonsense, tweets. Sure there is still nobody actively trying to solve the crisis. But at least by being checked out he won’t be undermining it either. So at least those who can help the situation might finally get traction. You know, the adults in the room. And that alone … him staying out of everyones way …will be the thing that could potentially save hundreds of thousands of peoples lives … regardless of mutations. At least until we get a true plan from a group that is competent.
I cannot save your articles for reading later. When your articles are saved, lot of unnecessary and unconnected picture stories at tehe end of the articles get printed. All,good publishers provide printable version for the articles. This is helpful and convenient for reader. Unnescessary material does not get downloaded due to this. Is it possible by you to provide this type of facility? I will appreciate it.
I don’t get any of this. A human tragedy. Nothing but complaints. The blame game. I’ve learned to relax n wait for my next transition. Death seems better than life here. Their has to be something better than this life. Hatred n greed rules the day! I am happy within myself. I have peace. And, dream each night of a wonderfully peaceful existence. Good luck to you all! Be assured that someone will greet you at the end!!
I was saying, months ago, look into the parasites tha burrow into humans.We have
The Helmiths,Bird and rat lice/mites. Flat mites soil mites etc. Some eat while they are being eaten by another. These have no nucleus I believe. Animals, they are.As such are humans.
,
The level of tinfoil hat idiocy demonstrated in the comment section of an article about covid-19 mutations is stunning. It’s the descent into conspiracy theories, baseless accusations, alienation of allies while fawning over demented tyrants and adversaries that flattered Trump’s ego, the complete reversion from critical thinking to cult like obsession is why I didn’t vote for Trump.
I’ve never seen a man more willing to harm our country for no other reason than to try push a bizarre narrative he is infallible and has done the best at everything alway. With Trump and his cult, if anything seems bad, is someone else’s fault. I’ve never seen a man more willing to throw anyone under the bus to preserve himself than Trump. But I can handle that Trump is who he is. His well documented history of deceit, narcissism, division, attacks and posturing goes back decades.
What keeps me up at night are the people who support him, the mindless lemmings who angrily, almost frantically, struggle to parrot as many conspiracies and include as many narratives the president has baselessly pushed into their every post. To them, if information is flattering to Trump, it’s the truest true they’ve ever heard. If it’s critical or contradicts something Trump has made up, it’s the evil machinations of the fake news, the socialists, the southern hordes, the qanon-revealed pedophile devil worshippers which apparently infect every Democrat in the world, . Information is accepted or rejected on whether it makes Trump look good. Trumpian conservatives have lost the ability to critically assess information. No evidence is needed, no reason is needed, just angrily complain about the socialist threat blah blah blah and maintaining that any election in which Trump doesn’t win must be fraudulent.
Right? No! Wrong! You’ve lost your minds and have infected an entire generation with a manner of thinking last popular in the dark ages.
Republicans keep complaining that people didn’t vote for biden, they voted against Trump (when they acknowledge that their votes are valid at all). You might be right – and it’s because Trump is terrible. Truly, astonishingly, terrible. Just awful. A terrible human being. A sexual predator, draft dodger, liar, cheat, criminal, narcissist, incompetent blowhard and idiot.
People don’t hate him for no reason, or because he’s just so great, or because they’re sore losers, although the irony of that last one is palpable at the moment, they hate him because he’s a terrible human being.
He’s a corrosive narcissist who has done generational damage to our country, our foreign relations, our unity, our brain trust and expertise, our faith in democrac; even now flailing around like a pathetic crybaby, determined to burn down our democracy on his way out the door.
It’s for these and COUNTLESS other reasons people voted against him. For those of you who voted for him, you have been conned like few groups in history. You’ve been played for fools via the one thing Trump is truly exceptional at: deceit. You’re not a movement; you’re a cult. and thank God you were not able to take yet another election with a minority of the vote. You’ve become entitled, angry children who refuse to accept personal responsibility and blame everything on anyone else, completely forgoing reason or evidence. This is Trump’s legacy: an anti science cult defined by its hypocrisy, hatred, irrationality and blame. The worst instincts of human beings were weaponized by donald Trump, and it’s time to start returning to sanity. Otherwise America will be over, and it will be at the hands of the Trump cult.
Hi Jane, Brian Tracey in the Psychology of Achievement said,”You cannot bring a negative person up, they want to be where they are and will only bring you down.” Self responsibility is the path to maturity & good mental health.
Hahahahahahahahaha!!! Another scientific talk ran over peoples political OPINION! OPINIONS are like your lower orifice, you all have one and they All stink. Keep your opinion where they belong. This is science. And yet noted many incorrect statements. All living entities on Earth used DNA in a form of nucleus or nucleolus. However, virus have single strand or double stranded DNA or RNA.
I wonder at the upended fear in denial. People have died all over the world. My hope is for a vaccine asap so schools, churches and other places of community may safely open. Oh yeah, like Hillary, the Trump issue is another moot discussion, as is climate change..too late, the iceberg has fallen. Viral mutation may just take the edge off, so pray for that& accept the gifts from science as the lifesaving source.
When the VERY FIRST SENTENCE in an article IS FALSE, the entire article should be questioned.
The first documented case of COVID-19 in the US was in Washington state in January 2020, not in March. 🤦🏼♀️
“Before COVID-19 first entered the United States in March, Houston Methodist Hospital had already begun preparations to test for and sequence the virus on a large scale, given the news coming out of Wuhan, China.
Between March 5, when the first case turned up in metropolitan Houston”
I’m sorry I can’t take anything seriously from someone who can’t get this one simple fact right. The first known case of COVID in the U.S. was on January 20th, 2020 in Washington State, this has long been common knowledge.
So what? ALL retroviruses mutate. That’s why the vaccine thing is just a big lie and a total scam by the drug companies, same as the regular annual flu-vaccines they peddle every year that do nothing. An effective vaccine can’t be made for retroviruses, otherwise there would be a vaccination for AIDS by now. Just more ridiculous alarmist propaganda courtesy of the social and news medias about something that we can’t do a thing about – that’s all it is.