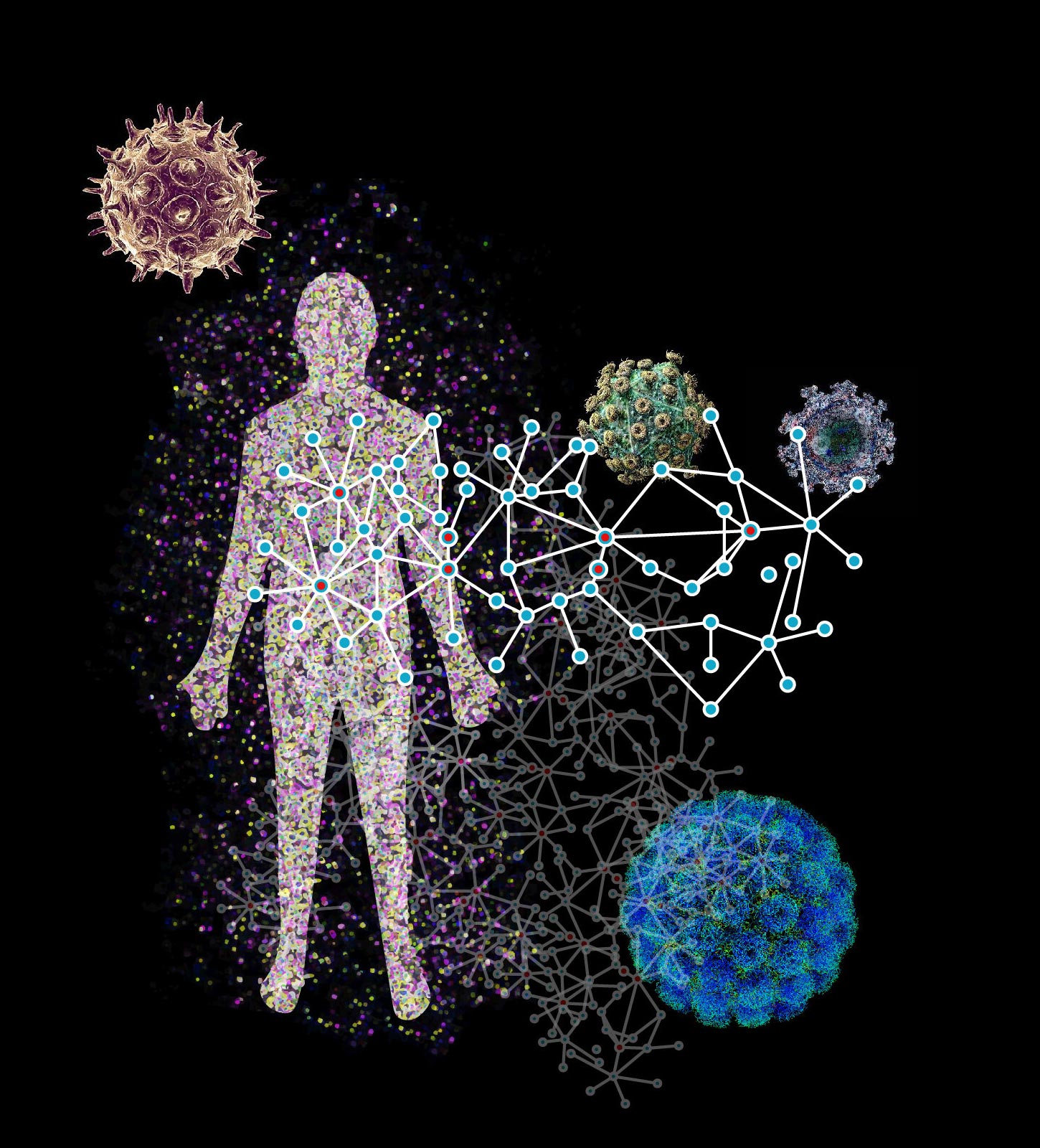
P-HIPSTer is a computational framework developed at Columbia University that infers interactions between pathogens and human proteins–the building blocks of viruses and cells. The framework is being used to understand how viruses infect human cells. Credit: Sagi Shapira, Ph.D., Columbia University Vagelos College of Physicians and Surgeons
Biologists at Columbia University Vagelos College of Physicians and Surgeons have leveraged a computational method to map protein-protein interactions between all known human-infecting viruses and the cells they infect. The method, along with the data that it generated, has generated a wealth of information about how viruses manipulate the cells that they infect and cause disease. Among the study’s findings are the role of estrogen receptors in regulating Zika virus infection and how human papillomavirus (HPV) causes cancer.
The study, led by Sagi Shapira, Ph.D., assistant professor of systems biology at Columbia University Vagelos College of Physicians and Surgeons, was published today in the journal Cell.
Limited Understanding of How Viruses Work
At the molecular level, viruses invade cells and manipulate them to replicate, survive, and cause disease. Since they depend on human cells for their life cycle, one-way viruses co-opt cellular machinery is through protein-protein interactions within their cell host. Similarly, cells respond to infection by initiating immune responses that control and limit viral replication – these too, depend on protein-protein interactions.
To date, considerable effort has been invested in identifying these key interactions – and many of these efforts have resulted in many fundamental discoveries, some with therapeutic implications. However, traditional methods are limited in terms of scalability, efficiency, and even access. To address this challenge, Dr. Shapira and his collaborators developed and implemented a computational framework, P-HIPSTER, that infers interactions between pathogen and human proteins–the building blocks of viruses and cells.
Until now, our knowledge about many viruses that infect people is limited to their genome sequences. Yet for most viruses, little has been uncovered about the underlying biological interactions that drive these relationships and give rise to disease.
“There are over 1,000 unique viruses that are known to infect people,” says Dr. Shapira. “Yet, despite their unquestionable public health importance, we know virtually nothing about the vast majority of them. We just know they infect human cells. The idea behind this effort was to systematically catalog the interactions that viruses have with the cells they infect. And, by doing so, also reveal some really interesting biology and provide the scientific community with a resource that they can use to make interesting observations of their own.”
Using a novel algorithm, P-HIPSTer exploits protein structural information to systematically interrogate virus-human protein-protein interactions with remarkable accuracy. Dr. Shapira and his collaborators applied P-HIPSTer to all 1,001 human-infecting viruses and the approximately 13,000 proteins they encode. The algorithm predicted roughly 280,000 likely pairs of interacting proteins that represent a comprehensive catalog of human virus protein-protein interactions with an accuracy rate of almost 80 percent.
“This is the first step towards building a comprehensive cartography of physical interactions between different organisms,” Dr. Shapira says.
Zika, HPV, Viral Evolution
In addition to defining pan-viral protein interactions, P-HIPSTer has yielded new biological insights into the Zika virus, HPV, and the impact of viruses in shaping human genetics.
Among their discoveries, the researchers found that the Zika virus interacts with the estrogen receptor, the protein that allows cells to effectively respond to the estrogen hormone. Importantly, they found estrogen receptor has the potential to inhibit Zika virus replication. Says Dr. Shapira, “And, in fact, estrogen receptor inhibits viral replication even more than interferon, a protein that is the body’s first line of defense to viral infection and our gold standard for anti-viral defense.”
The finding is particularly relevant to clinical disease as pregnant women are most susceptible to Zika during their first trimester, which is when estrogen levels are at their lowest. This period also is when the fetus is most susceptible to Zika, a virus for which there is no vaccine or specific treatment and that can cause severe birth defects.
Dr. Shapira and his team also explored interactions between HPV, the leading cause of cervical cancer, and the cells that it infects. HPV is the most common sexually transmitted viral infection with approximately 80 percent of sexually active individuals contracting one of the 200 different types of HPV at some point in their lives. Dr. Shapira and his team used the data generated by P-HIPSTer to identify protein-protein interactions that distinguish HPV infections associated with cancer from those that are not. In addition to providing insights into how HPV may cause disease, the finding could lead to improved diagnostics for those infected with HPV, and P-HIPSTer could potentially be used to help predict whether or not any particular virus is likely to be highly pathogenic.
The researchers also examined whether the interactions mediated by viruses have impacted human genetics. The researchers found evidence of strong selection pressure for several dozen cellular proteins that have been shaped by viral infection, unlocking new insights into how our genome has been impacted by viruses.
“One of the things we can do with this data is drilled down and ask whether virus infection has changed the history of human genetics,” notes Dr. Shapira. “That is certainly not a novel idea but to have a catalog of what those proteins are is significant. There are a lot of areas that we can explore now that we couldn’t before.”
Future Work
Dr. Shapira and his team intend to apply P-HIPSTer on more complex pathogens, such as parasites and bacteria, and use it to better understand how bacteria in the human gut communicate with each other. In the future, the algorithm could also be used to explore viruses or pathogens that affect agricultural plants or livestock.
The Shapira Laboratory at Columbia University is working to decipher the genetic and molecular circuitry at the interface of host-pathogen interactions. A deeper understanding of these relationships provides important insights into cellular machinery that control basic cell biology and has broad implications in human translational immunology and infectious disease research.
###
Reference: “A Structure-Informed Atlas of Human-Virus Interactions” by Gorka Lasso, Sandra V. Mayer, Evandro R. Winkelmann, Tim Chu, Oliver Elliot, Juan Angel Patino-Galindo, Kernyu Park, Raul Rabadan, Barry Honig, Sagi D. Shapira, 29 August 2019, Cell.
DOI: 10.1016/j.cell.2019.08.005
Summary
While knowledge of protein-protein interactions (PPIs) is critical for understanding virus-host relationships, limitations on the scalability of high-throughput methods have hampered their identification beyond a number of well-studied viruses. Here, we implement an in silico computational framework (pathogen host interactome prediction using structure similarity [P-HIPSTer]) that employs structural information to predict ∼282,000 pan viral-human PPIs with an experimental validation rate of ∼76%. In addition to rediscovering known biology, P-HIPSTer has yielded a series of new findings: the discovery of shared and unique machinery employed across human-infecting viruses, a likely role for ZIKV-ESR1 interactions in modulating viral replication, the identification of PPIs that discriminate between human papilloma viruses (HPVs) with high and low oncogenic potential, and a structure-enabled history of evolutionary selective pressure imposed on the human proteome. Further, P-HIPSTer enables discovery of previously unappreciated cellular circuits that act on human-infecting viruses and provides insight into experimentally intractable viruses.


Be the first to comment on "How Viruses Infect Humans Shown in Detailed Map"