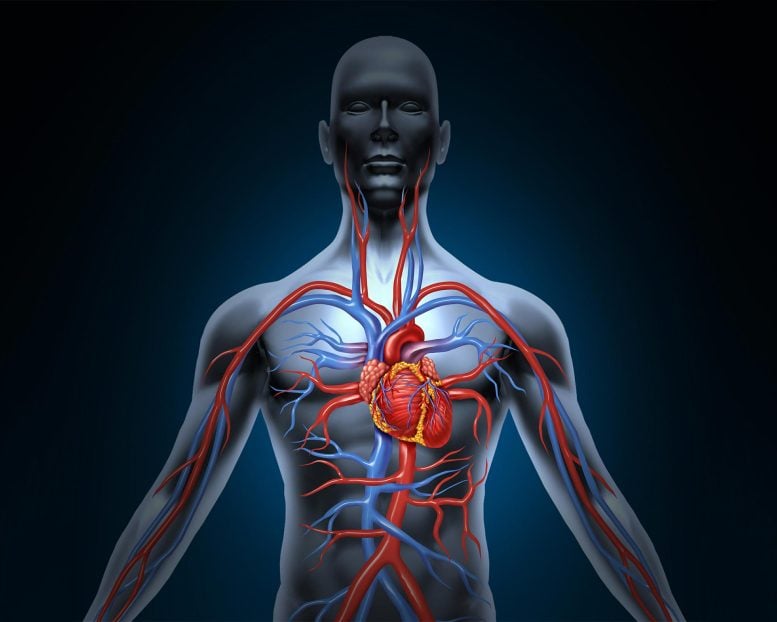
Pulmonary arterial hypertension (PAH) is a condition resulting from elevated blood pressure in the pulmonary blood vessels. Impairment of a critical signaling cascade in these vessels plays a significant role in PAH.
A newly published study from Yale University scientists could lead to the development of new therapies aimed at restoring the signaling balance in the pulmonary vessels in order to treat patients with pulmonary arterial hypertension.
Impairment of a key signaling cascade in the pulmonary blood vessels plays an important role in pulmonary arterial hypertension, a Yale study has found. The study appears in the advance online publication of Nature Medicine.
Pulmonary arterial hypertension (PAH) is a disease caused by an increase of blood pressure in the blood vessels of the lungs. If untreated, the majority of patients with the disease will succumb to heart failure and death.
PAH is characterized by the formation of lesions in the lungs composed of abnormally proliferating cells of the endothelium (cell tissue that lines blood vessels and the heart) and vascular smooth muscle cells. Recent studies have described the role of the gene apelin in the signaling process that maintains normal pulmonary vascular function. Apelin levels have been found to be significantly reduced in patients with PAH.
The Yale team set out to discover the mechanism by which impaired apelin signaling triggers the abnormal proliferation of cells that characterize PAH. They focused on the connection between reduced apelin expression and increased fibroblast growth factor (FGF) signaling. FGF helps to form and maintain blood vessels when functioning normally, but if produced in excess, as it is in diseases such as PAH, it can cause abnormal proliferation of cells, leading to pathologic remodeling of blood vessels.
In studies of rodent and human tissue samples, the scientists found that increased expression of two key components of FGF signaling, FGF2 and its receptor FGFR1, resulted from decreased levels of two microRNAs that are regulated by apelin. Decreased expression of these microRNAs disrupted the pulmonary vascular system’s ability to maintain cellular balance, resulting in abnormal induction of cellular growth in PAH. The scientists found that restoring these microRNAs in rat models of PAH led to dramatic reversal of the disease.
“Our findings could lead to development of new therapies aimed at restoring the signaling balance in the pulmonary vessels in order to treat patients with pulmonary arterial hypertension,” said senior author Dr. Hyung Chun, assistant professor of medicine (cardiology) at Yale School of Medicine. “New treatments are critical, as close to half of the patients diagnosed with PAH die within three years despite currently available therapies.”
Reference: “An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension” by Jongmin Kim, Yujung Kang, Yoko Kojima, Janet K Lighthouse, Xiaoyue Hu, Micheala A Aldred, Danielle L McLean, Hyekyung Park, Suzy A Comhair, Daniel M Greif, Serpil C Erzurum and Hyung J Chun, 23 December 2012, Nature Medicine.
DOI: 10.1038/nm.3040
Other authors are Jongmin Kim, Yujung Kang, Yoko Kojima, Janet Lighthouse, Xiaoyue Hu, Danielle McLean, Hyekyung Park and Daniel Greif of Yale School of Medicine; Micheala Aldred, Suzy Comhair, and Serpil Erzurum of the Lerner Institute, Cleveland Clinic Foundation.
The study was supported by grants from National Institutes of Health (HL095654, HL11005, HL101284, HL069170, and HL093362), the Howard Hughes Medical Institute Physician Scientist Early Career Award, the American Heart Association Grant-in-Aid, and the Pfizer ASPIRE Young Investigator Research Award.






We are all hoping that there time will come where almost all disease and disorder can be treated. Though we can see scientist and researchers are doing their best to have a good treatment to certain diseases. Hypertension is not an easy condition, if we disregard it, it can cause death.
As a pah patient I would love to know where this study stands and if they have plans to move forward with a prescription
Agree with Cindy…It would be interesting to see a status report on the efforts to move forward with prescription.