Surprisingly, certain patients with the mutated PDE3A gene were immune to hypertension-related damage.
Scientists in Berlin have been studying a strange hereditary condition that causes half the people in certain families to have shockingly short fingers and abnormally high blood pressure for decades. If untreated, affected individuals often die of a stroke at the age of 50. Researchers at the Max Delbrück Center (MDC) in Berlin discovered the origin of the condition in 2015 and were able to verify it five years later using animal models: a mutation in the phosphodiesterase 3A gene (PDE3A) causes its encoded enzyme to become overactive, altering bone growth and causing blood vessel hyperplasia, resulting in high blood pressure.
Immune to Hypertension-Related Damage
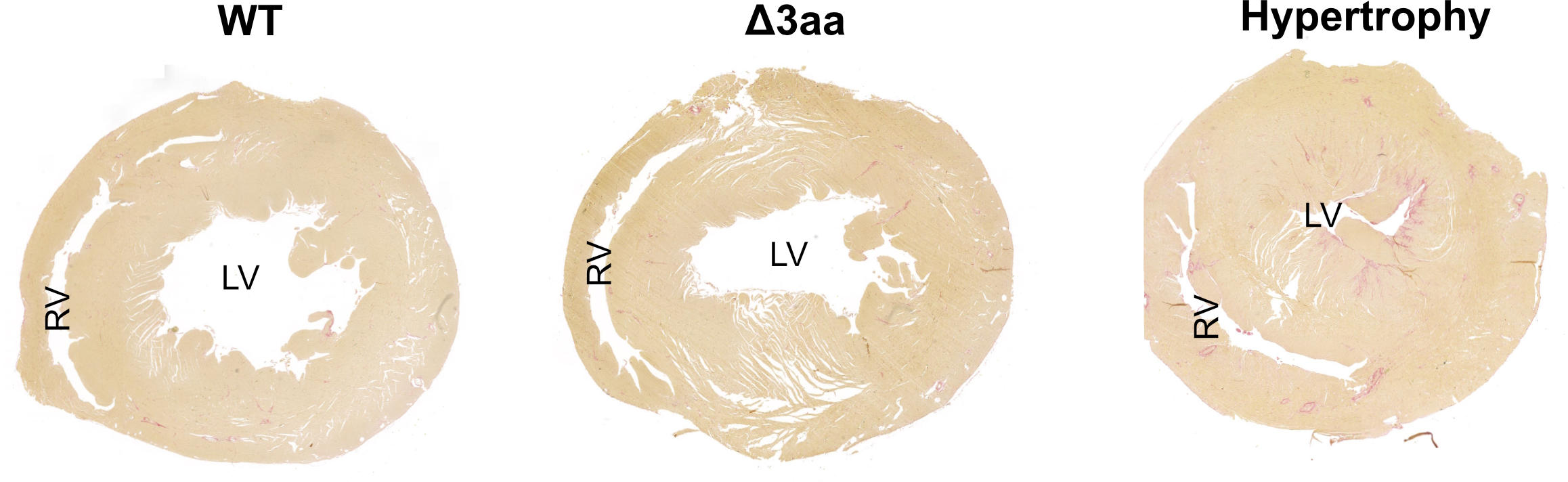
“High blood pressure almost always leads to the heart becoming weaker,” says Dr. Enno Klußmann, head of the Anchored Signaling Lab at the Max Delbrück Center and a scientist at the German Centre for Cardiovascular Research (DZHK). As it has to pump against a higher pressure, Klußmann explains, the organ tries to strengthen its left ventricle. “But ultimately, this results in the thickening of the heart muscle – known as cardiac hypertrophy – which can lead to heart failure greatly decreasing its pumping capacity.”
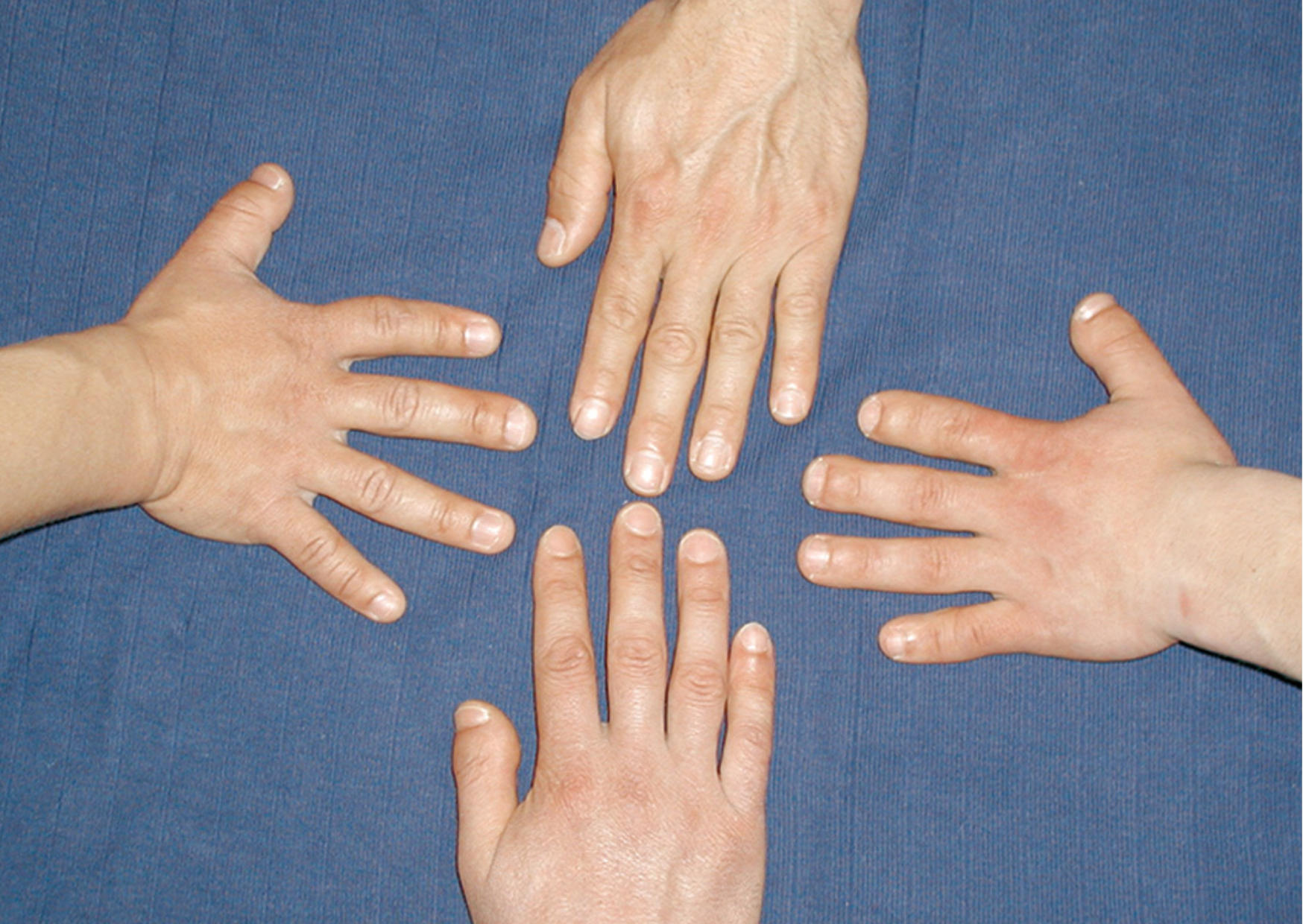
However, this doesn’t happen in hypertension patients with short fingers and mutant PDE3A genes. “For reasons that are now partly – but not yet fully – understood, their hearts appear immune to the damage that usually results from high blood pressure,” says Klußmann.
The research was conducted by scientists from the Max Delbrück Center, Charité – Universitätsmedizin Berlin, and the DZHK and has been published in the journal Circulation. In addition to Klußmann, final authors included Max Delbrück Center professors Norbert Hübner and Michael Bader, as well as Dr. Sylvia Bähring from the Experimental and Clinical Research Center (ECRC), a joint institution of Charité and the Max Delbrück Center.
The team, which included 43 other researchers from Berlin, Bochum, Heidelberg, Kassel, Limburg, Lübeck, Canada, and New Zealand, has recently published their findings on the protective effects of the gene mutation – and why these discoveries might transform the way heart failure is treated in the future. The study has four first authors, three of which are Max Delbrück Center researchers and one at the ECRC.
Two Mutations With the Same Effect
The scientists performed their tests on human patients with hypertension and brachydactyly (HTNB) syndrome – i.e., high blood pressure and abnormally short digits – as well as on rat models and heart muscle cells. The cells were grown from specially engineered stem cells known as induced pluripotent stem cells. Before testing began, researchers altered the PDE3A gene in the cells and the animals to mimic HTNB mutations.
“We came across a previously unknown PDE3A gene mutation in the patients we examined,” reports Bähring. “Previous studies had always shown the mutation in the enzyme to be located outside the catalytic domain – but we have now found a mutation right in the center of this domain.” Surprisingly, both mutations have the same effect in that they make the enzyme more active than usual. This hyperactivity ramps up the degradation of one of the cell’s important signaling molecules known as cAMP (cyclic adenosine monophosphate), which is involved in the contraction of the heart muscle cells. “It is possible that this gene modification – regardless of its location – causes two or more PDE3A molecules to cluster together and thus work more effectively,” Bähring suspects.
The Proteins Stay the Same
The researchers used a rat model – created with CRISPR-Cas9 technology by Michael Bader’s lab at the Max Delbrück Center – to try to better understand the effects of the mutations. “We treated the animals with the agent isoproterenol, a so-called beta-receptor agonist,” says Klußmann. Such medications are sometimes used in patients with end-stage heart failure. Isoproterenol is known to induce cardiac hypertrophy. “Yet surprisingly, this occurred in the gene-modified rats in a manner similar to what we observed in the wild-type animals. Contrary to what we expected, the existing hypertension did not aggravate the situation,” reports Klußmann. “Their hearts were quite obviously protected from this effect of the isoproterenol.”
In further experiments, the team investigated whether proteins in a specific signaling cascade of the heart muscle cells changed as a result of the mutation and if so which ones. Through this chain of chemical reactions, the heart responds to adrenaline and beats faster in response to situations such as excitement. Adrenaline activates the cells’ beta receptors, causing them to produce more cAMP. PDE3A and other PDEs stop the process by chemically altering cAMP. “However, we found little difference between mutant and wild-type rats at both the protein and the RNA levels,” Klußmann says.
More Calcium in the Cytosol
The conversion of cAMP by PDE3A does not occur just anywhere in the heart muscle cell, but near a tubular membrane system that stores calcium ions. A release of these ions into the cytosol of the cell triggers muscle contraction, thus making the heartbeat. After the contraction, the calcium is pumped back into storage by a protein complex. This process is also regulated locally by PDE.
Klußmann and his team hypothesized that because these enzymes are hyperactive in the local region around the calcium pump, there should be less cAMP – which would inhibit the pump’s activity. “In the gene-modified heart muscle cells, we actually showed that the calcium ions remain in the cytosol longer than usual,” says Dr. Maria Ercu, a member of Klußmann’s lab and one of the study’s four first authors. “This could increase the contractile force of the cells.”
Activating Instead of Inhibiting
“PDE3 inhibitors are currently in use for acute heart failure treatment to increase cAMP levels,” Klußmann explains. Regular therapy with these drugs would rapidly sap the heart muscle’s strength. “Our findings now suggest that not the inhibition of PDE3, but – on the contrary – the selective activation of PDE3A may be a new and vastly improved approach for preventing and treating hypertension-induced cardiac damage like hypertrophic cardiomyopathy and heart failure,” Klußmann says.
But before that can happen, he says, more light needs to be shed on the protective effects of the mutation. “We have observed that PDE3A not only becomes more active, but also that its concentration in heart muscle cells decreases,” the researcher reports, adding that it is possible that the former can be explained by oligomerization – a mechanism that involves at least two enzyme molecules working together. “In this case,” says Klußmann, “we could probably develop strategies that artificially initiate local oligomerization – thus mimicking the protective effect for the heart.”
Reference: “Mutant Phosphodiesterase 3A Protects From Hypertension-Induced Cardiac Damage” by Maria Ercu, Michael B. Mücke, Tamara Pallien, Lajos Markó, Anastasiia Sholokh, Carolin Schächterle, Atakan Aydin, Alexa Kidd, Stephan Walter, Yasmin Esmati, Brandon J. McMurray, Daniella F. Lato, Daniele Yumi Sunaga-Franze, Philip H. Dierks, Barbara Isabel Montesinos Flores, Ryan Walker-Gray, Maolian Gong, Claudia Merticariu, Kerstin Zühlke, Michael Russwurm, Tiannan Liu, Theda U.P. Batolomaeus, Sabine Pautz, Stefanie Schelenz, Martin Taube, Hanna Napieczynska, Arnd Heuser, Jenny Eichhorst, Martin Lehmann, Duncan C. Miller, Sebastian Diecke, Fatimunnisa Qadri, Elena Popova, Reika Langanki, Matthew A. Movsesian, Friedrich W. Herberg, Sofia K. Forslund, Dominik N. Müller, Tatiana Borodina, Philipp G. Maass, Sylvia Bähring, Norbert Hübner, Michael Bader and Enno Klussmann, 19 October 2022, Circulation.
DOI: 10.1161/CIRCULATIONAHA.122.060210
Never miss a breakthrough: Join the SciTechDaily newsletter.
Follow us on Google and Google News.