
A Quantum Computational Solution for Engineering Materials
Researchers at Argonne explore the possibility of solving the electronic structures of complex molecules using a quantum computer.
If you know the atoms that compose a particular molecule or solid material, the interactions between those atoms can be determined computationally, by solving quantum mechanical equations — at least, if the molecule is small and simple. However, solving these equations, critical for fields from materials engineering to drug design, requires a prohibitively long computational time for complex molecules and materials.
Now, researchers at the U.S. Department of Energy’s (DOE) Argonne National Laboratory and the University of Chicago’s Pritzker School of Molecular Engineering (PME) and Department of Chemistry have explored the possibility of solving these electronic structures using a quantum computer.
“This is an exciting step toward using quantum computers to tackle challenging problems in computational chemistry.” Giulia Galli
The research, which uses a combination of new computational approaches, was published online in the Journal of Chemical Theory and Computation. It was supported by Q-NEXT, a DOE National Quantum Information Science Research Center led by Argonne, and by the Midwest Integrated Center for Computational Materials (MICCoM).
“This is an exciting step toward using quantum computers to tackle challenging problems in computational chemistry,” said Giulia Galli, who led the research with Marco Govoni, a staff scientist at Argonne and member of the UChicago Consortium for Advanced Science and Engineering (CASE).
A Computational Challenge
Predicting the electronic structure of a material involves solving complex equations that determine how electrons interact, as well as modeling how various possible structures compare to each other in their overall energy levels.
Unlike conventional computers that store information in binary bits, quantum computers use qubits that can exist in superposition of states, letting them solve certain problems more easily and quickly. Computational chemists have debated whether and when quantum computers might eventually be able to tackle the electronic structure problem of complex materials better than conventional computers. However, today’s quantum computers remain relatively small and produce noisy data.
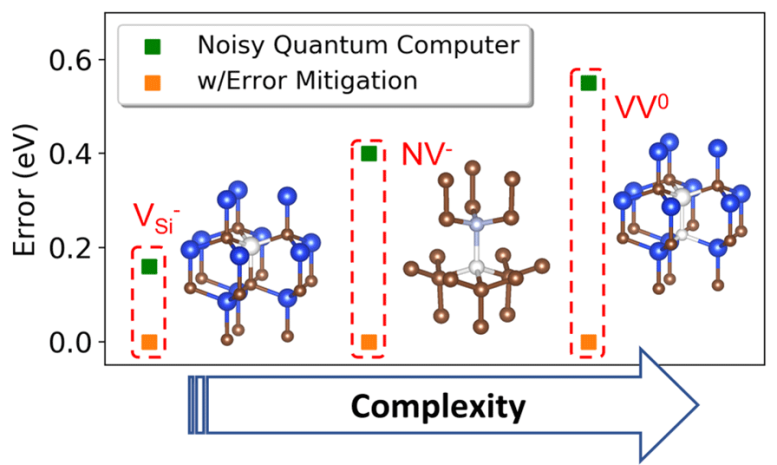
Even with these weaknesses, Galli and her colleagues wondered whether they still could make progress in creating the underlying quantum computational methods required to solve electronic structure problems on quantum computers.
“The question we really wanted to address is what is possible to do with the current state of quantum computers,” Govoni said. “We asked the question: Even if the results of quantum computers are noisy, can they still be useful to solve interesting problems in materials science?”
An Iterative Process
The researchers designed a hybrid simulation process, using IBM quantum computers. In their approach, a small number of qubits — between four and six — perform part of the calculations, and the results are then further processed using a classical computer.
“We designed an iterative computational process that takes advantages of the strengths of both quantum and conventional computers,” said Benchen Huang, a graduate student in the Galli Group and first author of the new paper.
After several iterations, the simulation process was able to provide the correct electronic structures of several spin defects in solid-state materials. In addition, the team developed a new error mitigation approach to help control for the inherent noise generated by the quantum computer and ensure accuracy of the results.
Hints at the Future
For now, the electronic structures solved using the new quantum computational approach could already be solved using a conventional computer. Therefore, the longstanding debate of whether a quantum computer can be superior to a classical one in solving electronic structure problems is not settled yet.
However, the results provided by the new method pave the way for quantum computers to address more complex chemical structures.
“When we scale this up to 100 qubits instead of 4 or 6, we think we might have an advantage over conventional computers,” Huang said. “But only time will tell for sure.”
The research group plans to keep improving and scaling up their approach, as well as using it to solve different types of electronic problems, such as molecules in the presence of solvents, and molecules and materials in excited states.
Reference: “Quantum Simulations of Fermionic Hamiltonians with Efficient Encoding and Ansatz Schemes” by Benchen Huang, Nan Sheng, Marco Govoni and Giulia Galli, 15 February 2023, Journal of Chemical Theory and Computation.
DOI: 10.1021/acs.jctc.2c01119
This work is supported by the U.S. Department of Energy National Quantum Information Science Research Centers as part of the Q-NEXT center and through the Midwest Integrated Center for Computational Materials (MICCoM). Headquartered at Argonne, MICCoM is funded by the DOE Office of Basic Energy Sciences.
Never miss a breakthrough: Join the SciTechDaily newsletter.
Follow us on Google and Google News.
2 Comments
I want some more of this information because I’m really interested in this website.
This is a interesting information that I’ve came across.