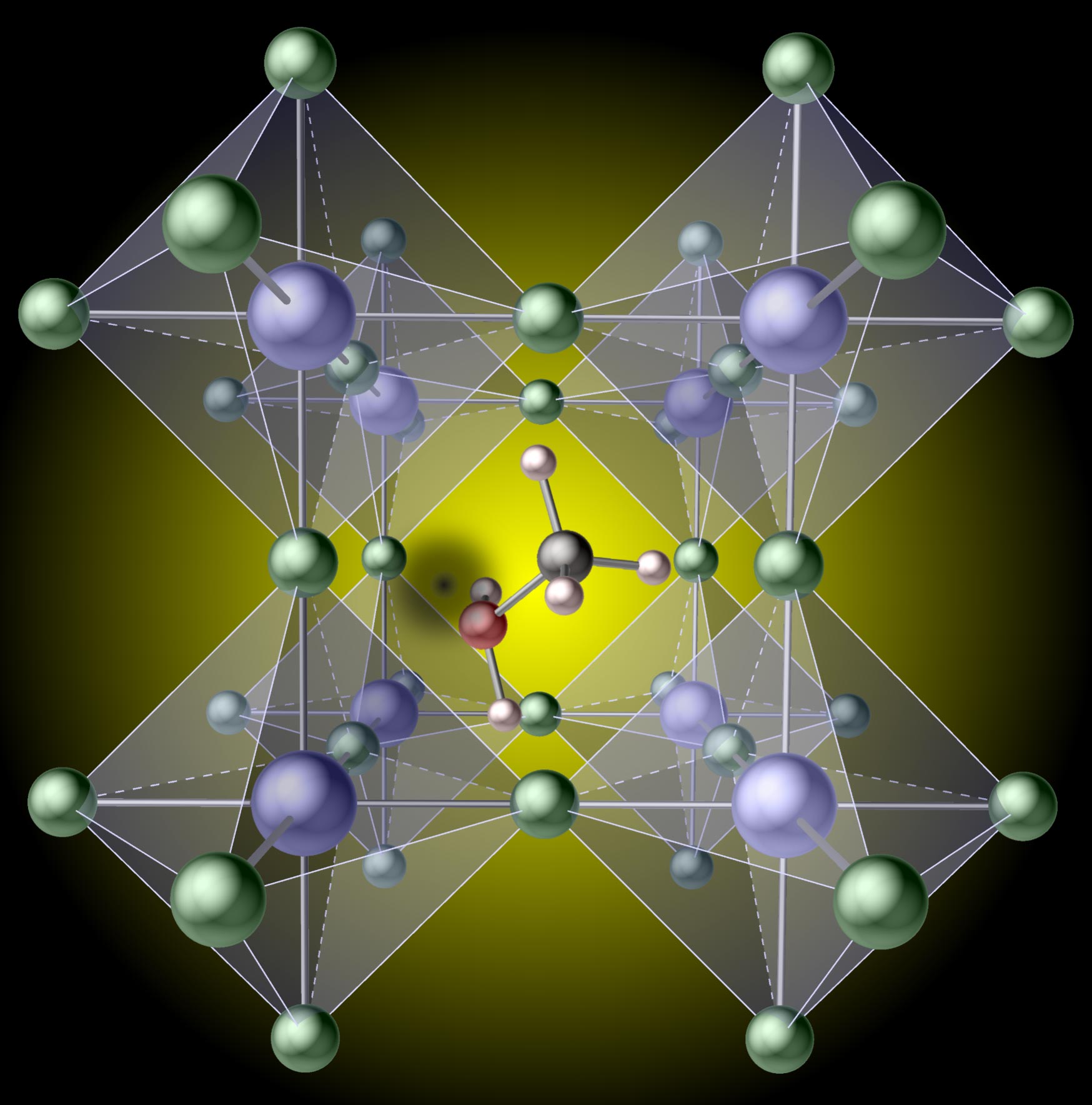
Missing hydrogen atoms in hybrid perovskites drastically reduce solar cell efficiency.
Researchers in the materials department in UC Santa Barbara’s College of Engineering have uncovered a major cause of limitations to efficiency in a new generation of solar cells.
Various possible defects in the lattice of what are known as hybrid perovskites had previously been considered as the potential cause of such limitations, but it was assumed that the organic molecules (the components responsible for the “hybrid” moniker) would remain intact. Cutting-edge computations have now revealed that missing hydrogen atoms in these molecules can cause massive efficiency losses. The findings are published in a paper titled “Minimizing hydrogen vacancies to enable highly efficient hybrid perovskites,” in the April 29 issue of the journal Nature Materials.
The remarkable photovoltaic performance of hybrid perovskites has created a great deal of excitement, given their potential to advance solar-cell technology. “Hybrid” refers to the embedding of organic molecules in an inorganic perovskite lattice, which has a crystal structure similar to that of the perovskite mineral (calcium titanium oxide). The materials exhibit power-conversion efficiencies rivaling that of silicon, but are much cheaper to produce. Defects in the perovskite crystalline lattice, however, are known to create unwanted energy dissipation in the form of heat, which limits efficiency.
A number of research teams have been studying such defects, among them the group of UCSB materials professor Chris Van de Walle, which recently achieved a breakthrough by discovering a detrimental defect in a place no one had looked before: on the organic molecule.

Hydrogen Vacancies
“Methylammonium lead iodide is the prototypical hybrid perovskite,” explained Xie Zhang, lead researcher on the project. “We found that it is surprisingly easy to break one of the bonds and remove a hydrogen atom on the methylammonium molecule. The resulting ‘hydrogen vacancy’ then acts as a sink for the electric charges that move through the crystal after being generated by light falling on the solar cell. When these charges get caught at the vacancy, they can no longer do useful work, such as charging a battery or powering a motor, hence the loss in efficiency.”
The research was enabled by advanced computational techniques developed by the Van de Walle group. Such state-of-the-art calculations provide detailed information about the quantum-mechanical behavior of electrons in the material. Mark Turiansky, a senior graduate student in Van de Walle’s group who was involved in the research, helped build sophisticated approaches for turning this information into quantitative values for rates of charge carrier trapping.
“Our group has created powerful methods for determining which processes cause efficiency loss,” Turiansky said, “and it is gratifying to see the approach provide such valuable insights for an important class of materials.”
Formamidinium Outperforms Methylammonium
“The computations act as a theoretical microscope that allows us to peer into the material with much higher resolution than can be achieved experimentally,” Van de Walle explained. “They also form a basis for rational materials design. Through trial and error, it has been found that perovskites in which the methylammonium molecule is replaced by formamidinium exhibit better performance. We are now able to attribute this improvement to the fact that hydrogen defects form less readily in the formamidinium compound.
“This insight provides a clear rationale for the empirically established wisdom that formamidinium is essential for realizing high-efficiency solar cells,” he added. “Based on these fundamental insights, the scientists who fabricate the materials can develop strategies to suppress the harmful defects, boosting additional efficiency enhancements in solar cells.”
Reference: “Minimizing hydrogen vacancies to enable highly efficient hybrid perovskites” by Xie Zhang, Jimmy-Xuan Shen, Mark E. Turiansky and Chris G. Van de Walle, 29 April 2021, Nature Materials.
DOI: 10.1038/s41563-021-00986-5
Funding for this research was provided by the Department of Energy’s Office of Science and Office of Basic Energy Sciences. The computations were performed at the National Energy Research Scientific Computing Center.
Never miss a breakthrough: Join the SciTechDaily newsletter.
Follow us on Google and Google News.