These slow growing bacteria have long puzzled TB researchers. Turns out the answer lies in the epigenetic domain.
For a slow-growing microbe that multiplies infrequently, Mycobacterium tuberculosis, the pathogen that causes tuberculosis (TB) has long puzzled researchers as to how it develops resistance to antibiotics so quickly, in a matter of weeks to months.
Now, TB researchers at San Diego State University have uncovered a crucial clue to the mystery: the answer may lie in the epigenetic domain rather than the genetic domain where most scientists have concentrated their efforts.
Their discovery could help advance new diagnostics, therapeutics, and vaccine targets.
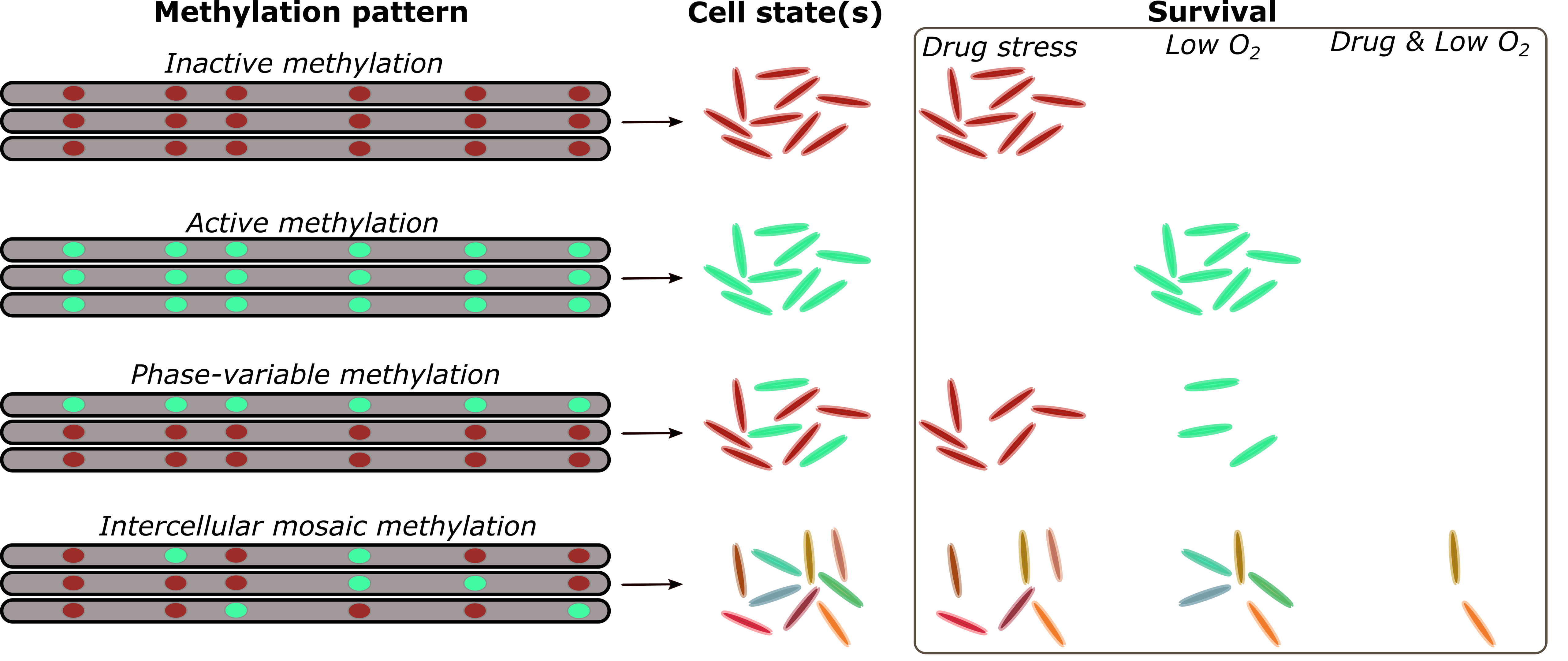
Epigenetics is the study of inheritable changes in gene expression that do not involve a corresponding change to the underlying DNA sequence — meaning changes to the phenotype but no change in the genotype. This affects only the physical structure of the DNA, through a process called DNA methylation where a chemical ‘cap’ is added to the DNA molecule, preventing or facilitating the expression of certain genes.
The SDSU researchers describe the rapid response phenomenon they discovered as ‘intercellular mosaic methylation,’ a process by which Mycobacterium tuberculosis diversifies, creating multiple subpopulations each with its own phenotype. While antibiotics could kill many of these mutant subpopulations, at least a few do survive and develop drug resistance.
“We believe this also explains why diagnostic testing in some patients does not predict treatment failure, and why some patients come back months later with the disease reemerging in a far more resistant state,” said Faramarz Valafar, a TB expert with SDSU’s School of Public Health who studies the genetics and epigenetics of pulmonary diseases. “This is also why CT scans of the lungs of many “cured” patients show lesions with possible bacterial activity.”
Worldwide, TB is among the top 10 causes of death. It killed 1.5 million people in 2018 and about 10 million people fall ill with it each year, according to the World Health Organization.
Valafar’s team collected hundreds of samples of drug-resistant varieties of the bacteria from patients in India, China, the Philippines, and South Africa, as well as Europe, through collaborations with TB researchers worldwide.
Their study was published in eLife in late October. Valafar and project scientist Samuel Modlin began exploring epigenetics for the TB bacterium in 2016, and doctoral student Derek Conkle-Gutierrez joined them in 2018, in the Laboratory for Pathogenesis of Clinical Drug Resistance and Persistence. Modlin, an SDSU alumnus, and Conkle-Gutierrez utilized skills and knowledge they acquired at SDSU to carry out this research — data and statistical analysis, coding skills, and bioinformatics knowledge.
“We’ve known for decades that bacterial epigenetics can influence the expression of certain genes, which can lead to a variety of phenotypes even when they have identical genotypes,” Conkle-Gutierrez said. “We discovered evidence of that phenomenon in the TB bacterium.”
Antibiotic resistance is typically caused by genomic mutations, but this bacterium is one of several that leverages alternative mechanisms in the epigenetic domain to enable rapid adaptation.
“We found that some of them had mutations that led to variable DNA methylation and those strains had much more diversity in their epigenome, and thus more potential to be drug resistant,” Modlin said.
The researchers found there were no set patterns and methylation was fairly random. They used advanced comparative genomic and epigenetic techniques to identify variations across cells within a colony from a single isolate, from a single patient — including tiny variations that nevertheless impacted gene expression. They were able to do this because, rather than assuming the reference genome has a common structure, they reconstructed each genome from scratch and analyzed its epigenetic signatures.
They will now focus on testing and confirming the key genes they identified with methylation signatures. There is more work to be done before their discovery can eventually be used for diagnostics.
“There is a lot of resistance in TB that escapes current molecular diagnostics and we don’t really know why. That’s problematic,” Valafar said. “This study offers a new domain, new tools, and a new approach to looking for alternative mechanisms. We move away from the classical view of molecular diagnostics and use a novel, comprehensive approach to analyzing bacteria.”
Current standard of care treatments use two types of antibiotics — bacteriostatics that prevent bacteria from multiplying but don’t kill them, and bactericides that do kill them.
“We found a new mode of variation and if we can inhibit that diversification mechanism, we can inhibit short-term epigenetic resistance and kill the bacteria before mutations in the genome develop and cause long-term, genetic resistance,” Modlin said.
This may be how some bacterial populations survive treatment and make the patient ill again with far greater antibiotic resistance or hypervirulence.
Reference: “Drivers and sites of diversity in the DNA adenine methylomes of 93 Mycobacterium tuberculosis complex clinical isolates” by Samuel J Modlin, Derek Conkle-Gutierrez, Calvin Kim, Scott N Mitchell, Christopher Morrissey, Brian C Weinrick, William R Jacobs, Sarah M Ramirez-Busby, Sven E Hoffner and Faramarz Valafar, 27 October 2020, eLife.
DOI: 10.7554/eLife.58542
The journal editors and reviewers consider their discovery to be significant and asked the researchers to rename three of the genes for which they describe new functions — MamC, MamS, MamS1. The article was also featured on the cover of the eLife magazine.
Valafar’s team has obtained two provisional patents for their discovery. The study was funded by the National Institute of Allergy and Infectious Diseases (NIAID).
Never miss a breakthrough: Join the SciTechDaily newsletter.
Follow us on Google and Google News.