Quantitative biologists David McCandlish and Juannan Zhou at Cold Spring Harbor Laboratory have developed an algorithm with predictive power, giving scientists the ability to see how specific genetic mutations can combine to make critical proteins change over the course of a species’ evolution.
Described in Nature Communications, the algorithm called “minimum epistasis interpolation” results in a visualization of how a protein could evolve to either become highly effective or not effective at all. They compared the functionality of thousands of versions of the protein, finding patterns in how mutations cause the protein to evolve from one functional form to another.
“Epistasis” describes any interaction between genetic mutations in which the effect of one gene is dependent upon the presence of another. In many cases, scientists assume that when reality does not align with their predictive models, these interactions between genes are at play. With this in mind, McCandlish created this new algorithm with the assumption that every mutation matters. The term “Interpolation” describes the act of predicting the evolutionary path of mutations a species might undergo to achieve optimal protein function.
The researchers created the algorithm by testing the effects of specific mutations occurring in the genes that make streptococcal GB1 protein. They chose the GB1 protein because of its complex structure, which would generate enormous numbers of possible mutations that could be combined in an enormous number of possible ways.
“Because of this complexity, visualization of this data set became so important,” says McCandlish. “We wanted to turn the numbers into a picture so that we can understand better what [the data] is telling us.”
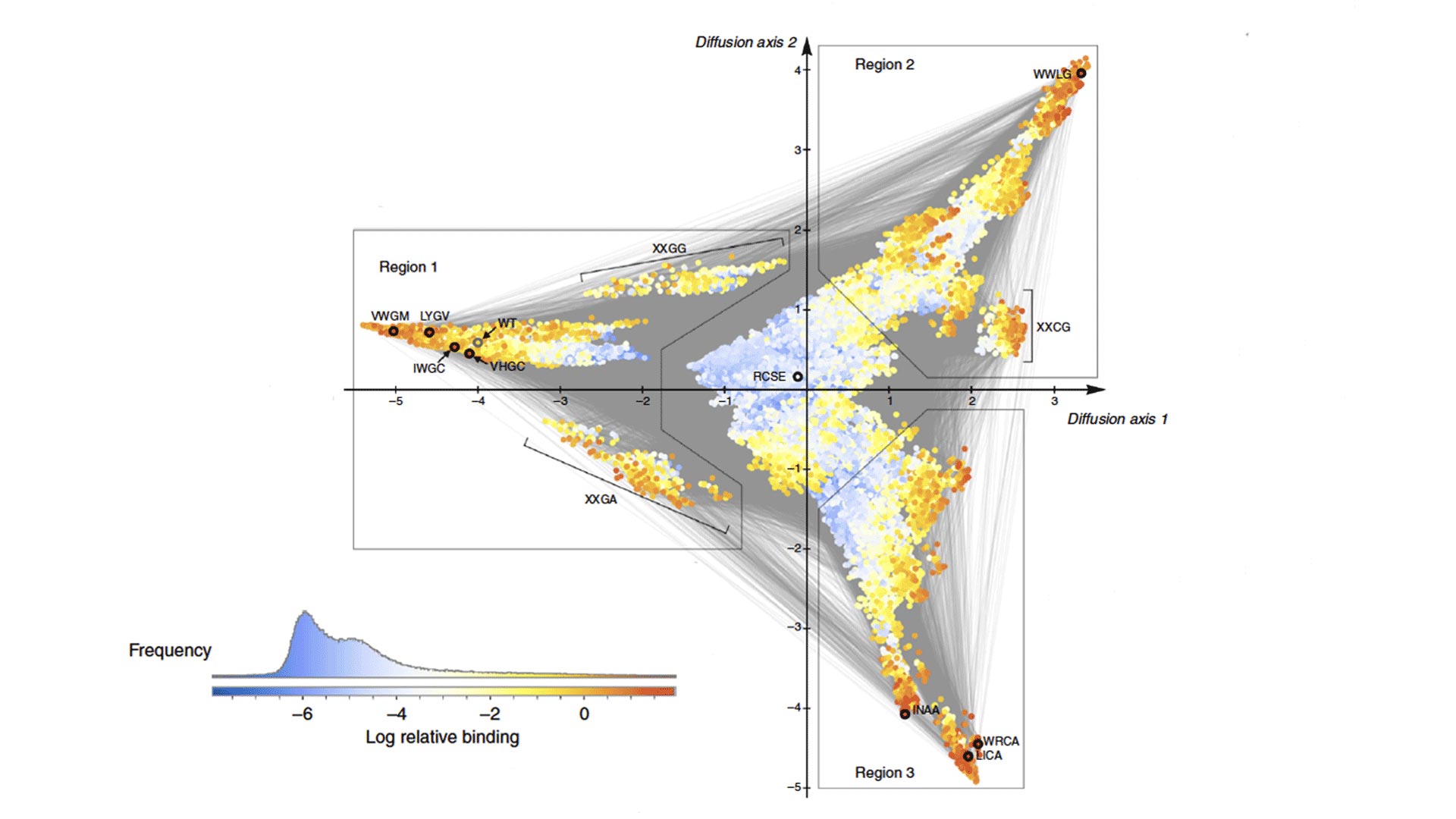
The visualization is like a topological map. Height and color correlate with the level of protein activity and distance between points on the map represents how long it takes for the mutations to evolve to that level of activity.
The GB1 protein begins in nature with a modest level of protein activity, but may evolve to a level of higher protein activity through a series of mutations that occur in several different places.

McCandlish likens the evolutionary path of the protein to hiking, where the protein is a hiker trying to get to the highest or best mountain peaks most efficiently. Genes evolve in the same manner: with a mutation seeking the path of least resistance and increased efficiency.
To get to the next best high peak in the mountain range, the hiker is more likely to travel along the ridgeline than hike all the way back down to the valley. Going along the ridgeline efficiently avoids another potentially tough ascent. In the visualization, the valley is the blue area, where combinations of mutations result in the lowest levels of protein activity.
The algorithm shows how optimal each possible mutant sequence is and how long it will take for one genetic sequence to mutate into any of many other possible sequences. The predictive power of the tool could prove particularly valuable in situations like the COVID-19 pandemic. Researchers need to know how a virus is evolving in order to know where and when to intercept it before it reaches its most dangerous form.
McCandlish explains that the algorithm can also help “understand the genetic routes that a virus might take as it evolves to evade the immune system or gain drug resistance. If we can understand the likely routes, then maybe we can design therapies that can prevent the evolution of resistance or immune evasion.”
There are additional potential applications for such a predictive genetic algorithm, including drug development and agriculture.
“You know, at the very beginning of genetics… there was all this interesting speculation as to what these genetic spaces would look like if you could actually look at them,” McCandlish added. “Now we’re really doing it! That’s really cool.”
Reference: “Minimum epistasis interpolation for sequence-function relationships” by Juannan Zhou and David M. McCandlish, 14 April 2020, Nature Communications.
DOI: 10.1038/s41467-020-15512-5
Never miss a breakthrough: Join the SciTechDaily newsletter.
Follow us on Google and Google News.