Root fungi can help or harm plants—depending on genes that control how aggressively they invade roots.
Complex microbial communities inhabit plants and modulate their development. Roots especially, host a wide diversity of micro-organisms – including bacteria and fungi – that directly influence plant health. Researchers from the MPIPZ previously discovered that these fungi are important members of the root microbiome that can promote plant growth, but only when they are kept in check by the combined action of the host innate immune system and root-inhabiting bacteria.
In a new study published in Nature Communications, Fantin Mesny, and co-authors provide novel insights into how these fungi colonize roots, why many of them are potentially harmful, and what differentiates beneficial from pathogenic fungi in the root mycobiome (i.e., the fungal component of the root microbiota).
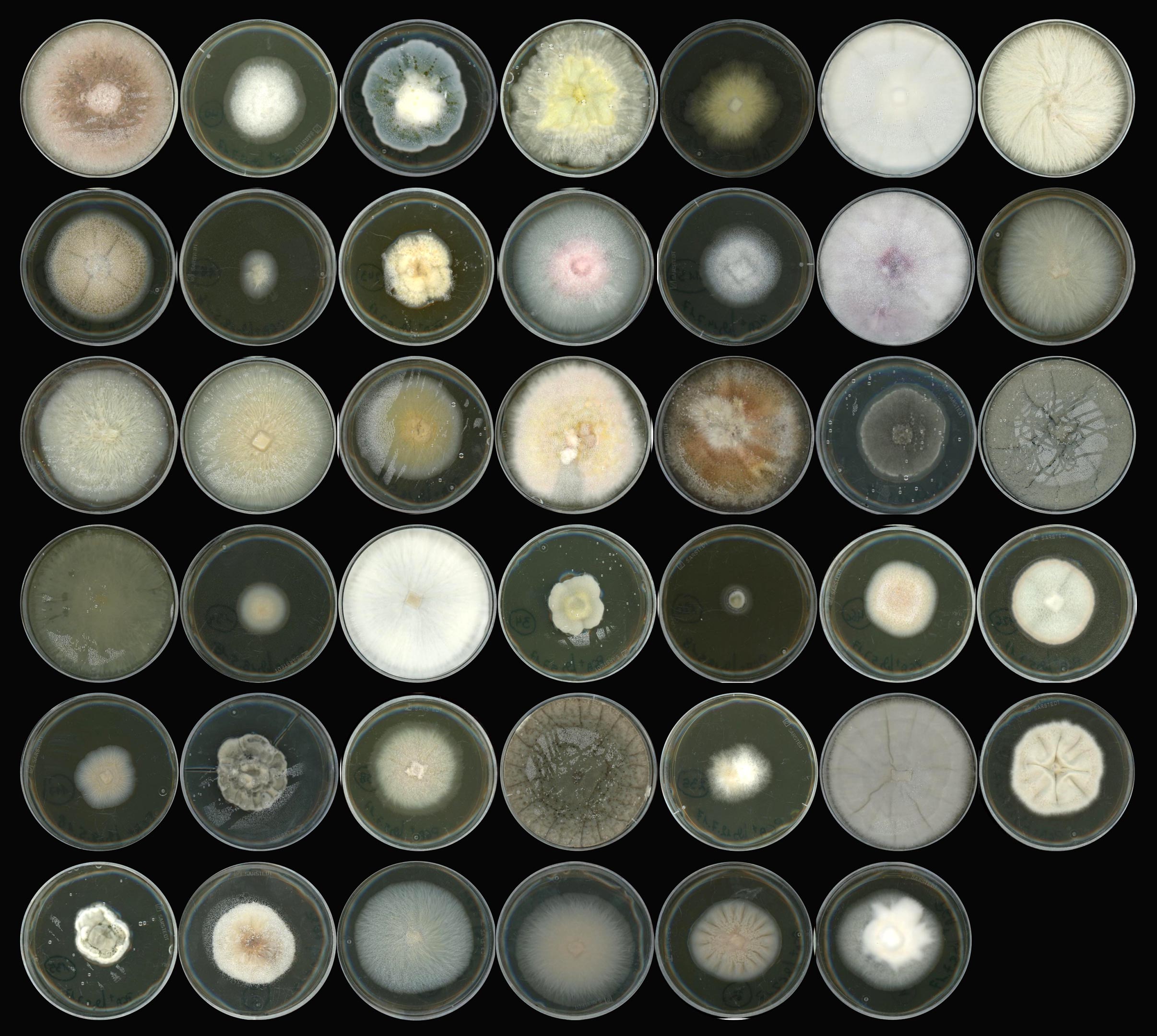
To address these questions, the researchers focused on the model plant Arabidopsis thaliana (Thale Cress), which cannot rely on beneficial mycorrhizal fungi to acquire nutrients since it does not harbor the genetic network needed to establish a functional symbiosis with these fungi. A. thaliana likely relies on other fungi to compensate for the loss of mycorrhizal partners and to survive in nature. To better characterize these root-colonizing fungi in their broad diversity, researchers have isolated a variety of fungal strains from the roots of healthy plants across Europe and selected 41 that are representative of the root mycobiome of A.thaliana (Figure 1).
Genomic Clues: Pathogenic Ancestry of Root Fungi
In collaboration with INRAE Nancy (France) and the JGI (USA), the genomes of these fungi were sequenced and compared to other fungi that were previously described as saprotrophic, pathogenic, endophytic, or mycorrhizal. Surprisingly, the scientists found that most root mycobiota members – isolated from the roots of healthy plants – derived from ancestors that were likely pathogenic, and have retained a battery of genes that were previously shown to be lost in genomes of beneficial mycorrhizal fungi. These genes encode effector-like small secreted proteins that could modulate the host immune system, and enzymes that can degrade a large number of plant cell-wall constituents including pectin, cellulose, and hemicellulose. These findings raised the possibility that many of these fungi may have retained at least part of their ancestral pathogenic capabilities.
To test this hypothesis, A. thaliana plants were grown in a closed system in the absence of any microorganism, or re-colonized with each of the selected 41 fungal isolates. This experiment identified a wide diversity of fungal effects on plant growth, ranging from highly detrimental to beneficial. Notably, the authors observed that the strains most harmful to the plant were colonizing roots much more aggressively than those having beneficial effects. Furthermore, the fungi most often detected in the roots of A. thaliana in nature were also the ones showing harmful effects on their host in mono-association experiments. Previous work from the group of Stéphane Hacquard had suggested that the mycobiome of A. thaliana can become detrimental when the host immune system and the root-inhabiting bacteria do not tightly control the proliferation of these fungi. These new results show that in nature, fungi with a high root-colonizing potential have a high pathogenic potential, explaining the need to control their growth.
Using a combination of association methods, including machine-learning models, the authors then associated the fungal effects on A. thaliana growth to genome compositions, and successfully identified a candidate gene family that could explain the detrimental effects and root colonization abilities. This family (pectate lyase PL1_7) encodes enzymes that degrade pectin, an essential constituent of plant cell walls, which is especially abundant in the roots of dicotyledonous plants such as A. thaliana. To validate its involvement in fungal detrimental activity, a gene from this family was introduced into the genome of a fungal species that naturally does not harbor it. The resulting mutant strain was able to colonize roots more aggressively than the original isolate and this increase in fungal load in roots was associated with a penalty on plant performance.
Cell-Wall Degrading Enzymes Drive Pathogenic Potential
According to the last author of the study Stéphane Hacquard, “These results indicate that repertoires of plant cell-wall degrading enzymes in fungal genomes are key genetic determinants driving access to the root endosphere and explaining why robust root colonizers can potentially become harmful if they degrade roots too aggressively.”
This study highlighted that the mycobiome of healthy plants in nature is composed of both friends and foes. This finding offers a new perspective on the effects of fungi on plant health, and possibly opens the door to new exciting considerations and developments for agriculture. Taking advantage of these results could potentially provide a rationale on how to design and optimize synthetic fungal communities to obtain beneficial outcomes on plant performance.
Reference: “Genetic determinants of endophytism in the Arabidopsis root mycobiome” by Fantin Mesny, Shingo Miyauchi, Thorsten Thiergart, Brigitte Pickel, Lea Atanasova, Magnus Karlsson, Bruno Hüttel, Kerrie W. Barry, Sajeet Haridas, Cindy Chen, Diane Bauer, William Andreopoulos, Jasmyn Pangilinan, Kurt LaButti, Robert Riley, Anna Lipzen, Alicia Clum, Elodie Drula, Bernard Henrissat, Annegret Kohler, Igor V. Grigoriev, Francis M. Martin and Stéphane Hacquard, 10 December 2021, Nature Communications.
DOI: 10.1038/s41467-021-27479-y
Never miss a breakthrough: Join the SciTechDaily newsletter.
Follow us on Google and Google News.