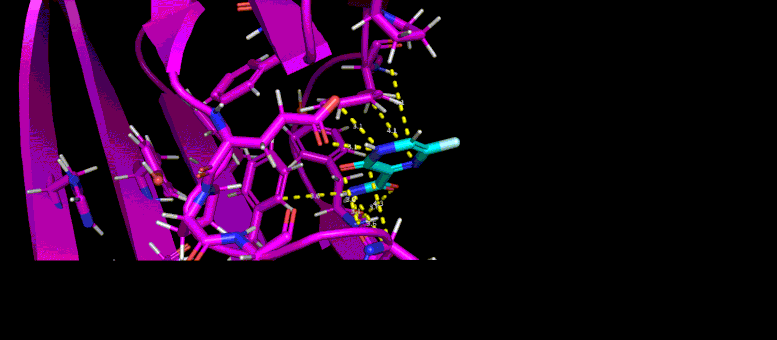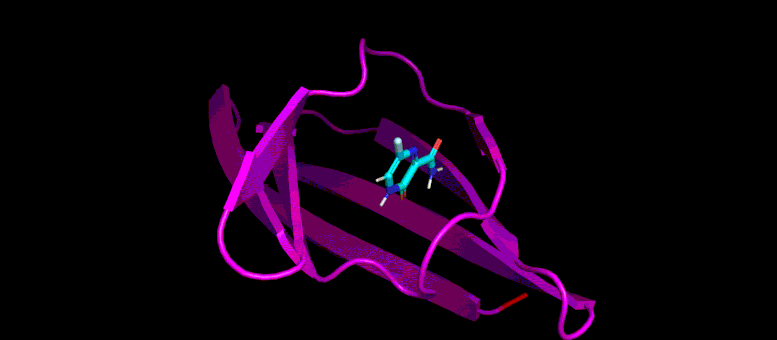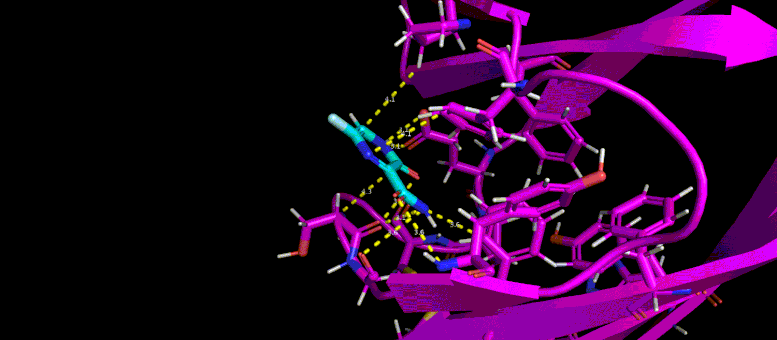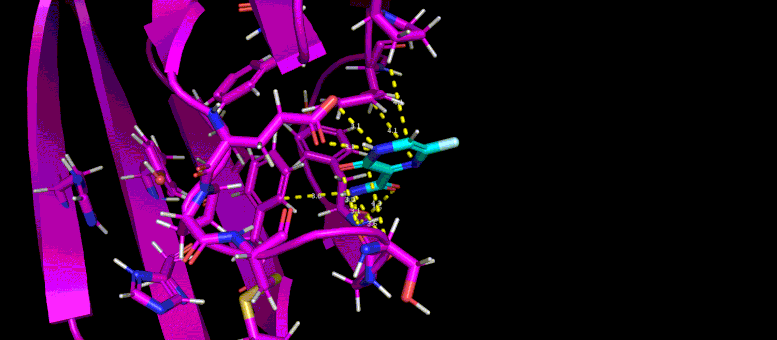
This movie of a 10-nanosecond molecular dynamics simulation shows how the shape of a SARS-CoV-2 virus protein (magenta) changes as it interacts with a possible small molecule inhibitor (blue).
Students conduct computational studies, explore inhibitor drugs to disrupt viral proteins that help infectious particles escape from cells.
Detailed knowledge of how SARS-CoV-2, the virus that causes COVID-19, replicates and how the body responds can point to different strategies for stopping it. Many researchers have been working to block the interaction of the coronavirus “spike” protein with the human-cell receptors to which it binds—the first step of infection. In contrast, three High School Research Program students participating in research with scientists in the Computational Science Initiative at the U.S. Department of Energy’s Brookhaven National Laboratory this summer took aim at one of the final steps—the virus’s exit strategy.
“After the virus is replicated and assembled, it needs to leave the cell,” explained Peggy Yin, a rising senior at Port Jefferson High School. “Our body has an immune response in the form of a protein called ‘tetherin’ that tethers the newly replicated virus particles to the cell membrane so they can’t get free to infect other cells. This is a really useful tactic that our body has built in to try to protect us.”
Unfortunately, SARS-CoV-2 has a way around this defense. The virus makes a protein that interferes with the tethering protein. “So maybe, if we inhibit the virus protein, we can let ‘tetherin’ do its thing,” Yin said.
The first step was to learn more about how the virus protein works.

Modeling molecular interactions
Yin and fellow HSRP students Jacob Zietek and Christopher Jannotta—who’d just graduated from Farmingdale and Eastport South Manor high schools, respectively—ran protein-protein docking studies to model how the viral protein and the tethering protein interact.
“Using this program, we can see where these proteins are talking to each other, where they’re binding to each other, and how the virus actually inhibits tetherin,” said Jannotta.
The modeling studies confirmed a suggestion the students had read about in the literature—that the virus protein binds to parts of the tether that get glycosylated (have sugar groups added), a necessary step for tetherin to work.
“We know that glycosylation happens in the endoplasmic reticulum, an internal organelle of the host cell,” Jannotta said. “That means if we were to develop some type of inhibitor of the virus protein, we might have to get it into this internal organelle. But at least now we knew where on the viral protein to look to dock possible inhibitors.”

Searching for inhibitors
The students conducted more docking studies—this time looking at the interactions of the virus protein with many possible small drug-like molecules, or “ligands,” to see which might work to stop the virus from blocking glycosylation.
“What these protein-ligand docking studies do is they try to fit the little ligands into the pocket that blocks glycosylation to find which one and which conformation, or ‘pose’ of the ligand, binds to the pocket best,” Yin said. From starting with 60 candidate ligands, the students narrowed the search to six or seven.
Then the team took the research even further by conducting molecular dynamics simulations of those candidate ligands. As Zietek explained, instead of just predicting whether a particular ligand will fit into a pocket on the protein—like a key fitting into a lock—molecular dynamics simulations predict how the shapes of the protein and ligand will conform to one another and change over time.
“These are much more complicated to compute,” Zietek said, because it gets down to what’s happening with individual atoms. “The program will calculate all the forces of atoms interacting with other atoms, and change the positions of the atoms relative to each other over time to match what would happen in real life as close as possible.”

Supercomputing power
“Because the project these students were working on is contributing to a broader collaboration—the National Virtual Biotechnology Laboratory (NVBL)—they had access to Brookhaven’s supercomputing clusters to run the molecular dynamics simulations,” said Hubertus Van Dam, their mentor.
That’s important because tracking the molecular interactions between each ligand and the viral protein for just 10 nanoseconds—10 billionths of a second—takes 8-12 hours to run even on such powerful machines. “On regular computers, it would take way too long!” Zietek noted.
Ten nanoseconds may not seem like a lot of time, but the simulations capture what happens every two femtoseconds—millionths of a billionth of a second, Jannotta noted. “The computer slows it down so we can see it in real time,” he said.
“It’s like a slow-motion camera,” Yin added.
As the team discovered by at first running even shorter simulations, 10 nanoseconds is long enough “to find out if a ligand will stick inside of a protein or if it’ll rip off,” Zietek said.
The students have quantified the results from the molecular dynamics simulations and are honing in on the best potential inhibitors, and identifying which pieces of the small drug-like molecules bind best to the virus protein. They are also looking at candidates to inhibit the viral protein in other ways. These initial computational studies pave the way for future experiments—and could even lead to other scientists, most likely at pharmaceutical companies, developing these ideas into actual drugs to inhibit SARS-CoV-2.
“I definitely think what we did is going to help all the scientists working on these therapeutic agents,” said Jannotta, who will begin biomedical engineering and premed studies at Stony Brook University this fall. “It’s going to help them get a more narrow view of what inhibitors might actually be worth pursuing in the pharmacology area. Other researchers maybe in pharmaceutical companies may take it up and carry it on. So I’m very excited for that.”
Virtual is reality
As Van Dam noted, “This is real-world research, with real potential impact. The fact that these internships ran ‘virtually’ really didn’t make a difference. Throughout the pandemic, even our professional computational scientists have been conducting similar computational studies working from our homes.”
Zietek, who will be attending Purdue University in the fall, said, “I never would have imagined that I’d be working on such a relevant and pressing topic as COVID-19 when I first applied for the Brookhaven program. I knew I wanted to work on a computational science project to learn more about how computers can be applied in a research setting. But this was just about the most emotional topic you could get for a project. I was very excited to get the opportunity to contribute.”
Yin was all in on the COVID angle from the start.
“In January, when I applied, I mentioned wanting to perform computational biology research in my essay. And as the pandemic situation worsened, I emailed about the possibility of me performing COVID-19 research, because I really wanted to help,” she said. “I’m hoping that since we know so little about coronaviruses in general, if somehow our research could shed light onto the mechanisms of how these viruses work, maybe this could help with other areas of coronavirus research and prevent other pandemics in the future.”
The CSI virus protein/drug-development modeling work is supported by the DOE Office of Science (BER) through the National Virtual Biotechnology Laboratory (NVBL), a consortium of DOE national laboratories focused on response to COVID-19, with funding provided by the Coronavirus CARES Act. Student participation in this project was supported through the HSRP, a program run by Brookhaven Lab’s Office of Educational Programs with funding from Brookhaven Science Associates—a partnership between Battelle and The Research Foundation for the State University of New York on behalf of Stony Brook University—which manages Brookhaven Lab.
Never miss a breakthrough: Join the SciTechDaily newsletter.
Follow us on Google and Google News.
2 Comments
Before you start writing an essay, you need to formulate an idea, define a goal, and find sources of information for your work. Use the free writing technique. Its essence is to write down all the thoughts that come to mind, without editing it and without following the grammar, punctuation, style, etc. A great way to help you cope with a creative crisis and find an extraordinary idea.
Thanks for sharing! Everyone wants to know when we will be able to freely leave the house, return to the office, walk as much as we want, meet with friends and have the opportunity to fly away on vacation to another country. For many students, time is important, the service https://cheappaperwriting.com/es/ will really help save time for such accomplishments.