In Brief
Problem: Development and trials of new coronavirus treatments may take anywhere from four to seven years. But millions of patients need treatment here and now. Doctors are looking for effective medications (or combinations of medicines), blindfolded. Meanwhile, chemists can predict which existing medications are potentially the most effective.
Solution: Russian researchers have created a special method of molecular modeling called ‘on-top docking.’ They used it to explore the whole surface of a protein that is vitally important for SARS-CoV-2 and compared it to a number of known medications. They discovered that two drugs are potentially able to ‘switch off’ the enzyme and stop the coronavirus reproduction. One of them is used to treat alcohol addiction, and the other is for cancer.
In Greater Detail
A team of chemists from HSE University and the Zelinsky Institute of Organic Chemistry used molecular modeling to find out that two medications that have been known for a long time can be used to fight SARS-CoV-2. These are disulfiram, which is used to treat alcoholism, and neratinib, an experimental drug being used to treat breast cancer. Both drugs are potential covalent inhibitors of the SARS-CoV-2 virus main protease Mpro – a key enzyme responsible for SARS-CoV-2 replication (copying its genetic material and building the new virus particles). The paper about the discovery was published in the July issue of Mendeleev Communications journal.

What Is This About?
A coronavirus was first detected in a patient with acute respiratory infection long ago, in 1965, but it was only about two decades ago that humanity faced really dangerous representatives of this family. Unfortunately, since the first SARS-CoV epidemic didn’t leave Asia (mostly, China) in 2002-2004 and the MERS outbreak in 2012-2015 seriously affected only Saudi Arabia and Korea, the global pharmaceutical industry has made virtually no effort to develop effective treatments for coronaviruses. Tests and medications have been actively developed almost exclusively for the needs of veterinary medicine.
Broad-spectrum medicines were used during previous epidemics, but the experience of medics in Chinese Wuhan demonstrated that this was not enough. Clinicians around the world have risked trying various experimental protocols, with the use of medicines used to treat HIV (lopinavir and ritonavir), malaria (chloroquine and hydroxychloroquine), and other diseases. But they were looking for the drugs being effectively blindfolded.
The global pharmaceutical industry was caught unaware, and there was no time to create brand new medicines. Even if potentially effective substances are detected, their preclinical and clinical trials would take from four to seven years. That’s why the most reasonable solution has been to search among known drugs that have proven to be safe for human health. This path – repurposing medicine – has been effectively used for a long time. The only problem is: how do we learn whether they are able to fight the coronavirus?
Computer modeling can help. This approach is called in silico — similarly to in vivo (in a living body) and in vitro (in a test tube). It allows numerical models to be used to test hundreds of various medications and determine their potential effectiveness and the mechanism of action. Chemists at HSE University and the RAS Zelinsky Institute of Organic Chemistry have been carrying out such research for many years. In 2014, they modeled a leukemia treatment, and in 2017, a treatment for rheumatoid arthritis. With such a background, the researchers jumped into the search for a SARS-CoV-2 treatment in 2020.
How Was it Studied?
The coronavirus, like many other viruses, mutates quite quickly. Its genome contains about 30,000 nucleotides – specific ‘building blocks’ of the genetic code. On average, one mutation, or more precisely, one SNP (single nucleotide polymorphism) happens in a virus RNA once every two weeks. This means that new strains of SARS-CoV-2 appear regularly. In Russia alone, there are nine unique SARS-CoV-2 lineages that are not present in other countries.
This is why the structural elements of the virus that are less subject to mutation during its evolution should be chosen as a target for the potential treatment. Otherwise, a medication effective against one strain would no longer be effective against another. The best candidates for this are conservative proteins, such as theSARS-CoV-2 virus main protease Mpro.In addition to being resistant to mutations, Mproplays a major role in coronavirus replication, which means that its inhibition (blocking its function) is able to slow down or even completely stop its reproduction inside the body.
Usually, the process of docking, as with a port dock and a ship entering it, is used for molecular modeling in simple cases. Two molecules participate in docking. One is called a ‘ligand’ (here, it is a medicine), and the other one is ‘receptor’ (or active site) of the target protein, such as Mpro, which can be used to ‘dock’. An effective drug docks with the active site, by covalent links, which makes the enzyme dysfunctional or destroys it.
To simulate the docking, researchers need to know the precise spatial structure of the drug molecule (they are available in special databases) and the precise configuration of the target protein’s active site. Here, researchers may face the first challenges: there might be dozens or even hundreds of such sites, and they are not fixed in space. That’s why classical docking does not work in SARS-CoV-2.
To overcome this problem, chemists from HSE University and the Zelinsky Institute decided to use ‘on-top docking’, which they came up with shortly before the pandemic. They decided not to focus on the previously described active site, but to investigate the whole surface of Mproprotein with many medications, hoping that the big calculation powers would return useful ‘dockings’.
The researchers used the spatial model ofSARS-CoV-2 Mprocreated in January 2020 from PDB database (ID 6LU7). The potential drugs were taken from the database of medications approved by the United States Food and Drug Administration (FDA). The research team’s own algorithms were used for modeling.
What Were the Results?
The modeling data demonstrated that sulfur-containing drugs show unusually high ligand efficiency at the active center of SARS-CoV-2 main protease Mpro, but only disulfiram 4 retains stable interactions.
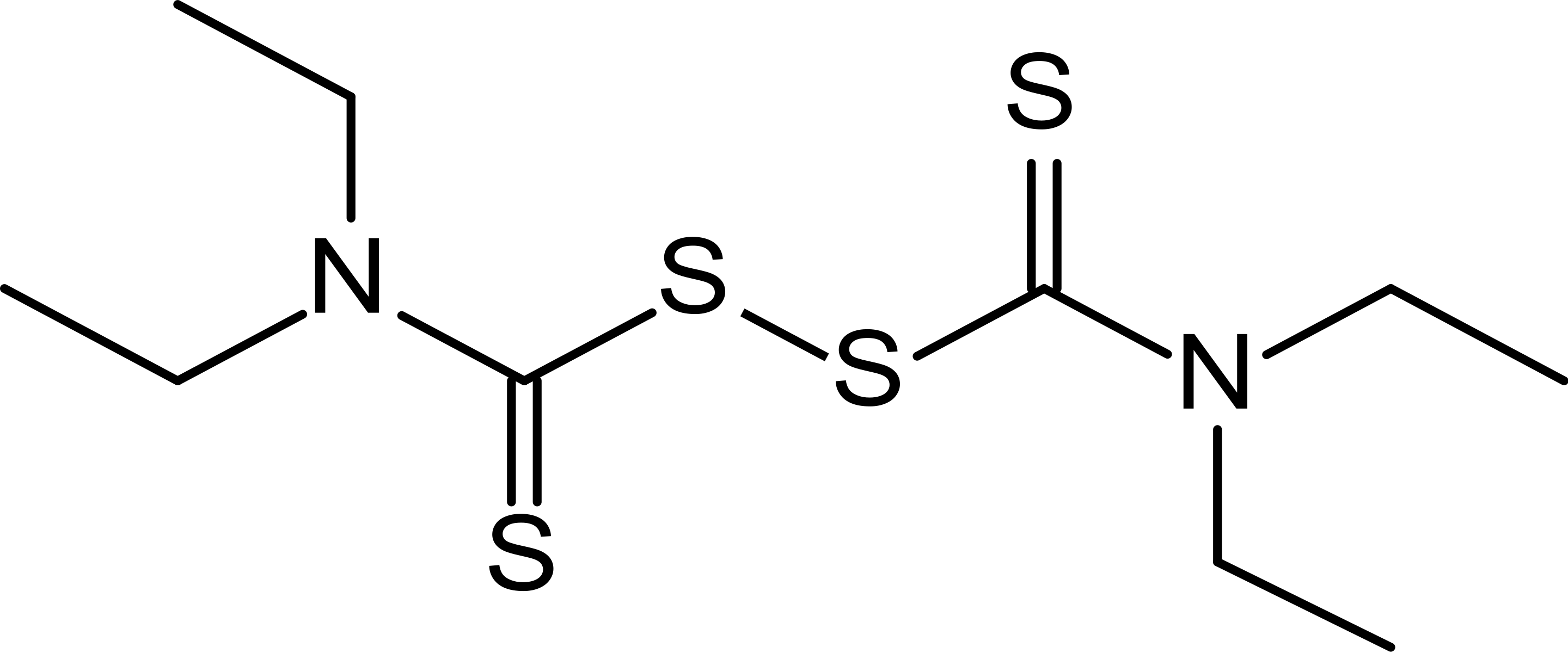
Today, it is most commonly used for treating alcoholism, since disulfiram inhibits the acetaldehyde dehydrogenase enzyme. As a result, the conversion of ethanol in the liver stops on the stage of acetaldehyde. Its concentration in the body grows, leading to acute intoxication, accompanied by sickness, vomiting and extreme pain. As a result, alcohol addicts acquire a conditioned reflex of disgust to the smell and taste of alcohol-containing drinks. This means that if the effectiveness of disulfiram against the novel coronavirus will be confirmed, this would help to solve two problems in Russia at once, at the same time decreasing alcohol addiction in the population.
Disulfiram fights SARS-CoV-2 in two ways. First, as previously demonstrated in vitro with SARS and MERS coronaviruses, it is a covalent inhibitor. In addition, it fights COVID-19 symptoms such as the significant decrease in reduced glutathione, which is an important antioxidant. This deficiency may lead to severe manifestations of the disease.
In addition to disulfiram, the Russian chemists were the first to predict the potential efficiency of neratinib, an irreversible tyrosine kinase inhibitor, against SARS-CoV-2. Just recently, in 2017, FDA approved neratinib as an adjuvant treatment of breast cancer.
How Can This Be Used?
Modeling has shown that both potential inhibitors of the main coronavirus protease (Mpro) are, presumably, covalent. For example, disulfiram can probably block the Mpro enzymatic activity by thiol–disulfide exchange reaction, while neratinib binding suggests the possibility of covalent interaction similarly to covalent peptide inhibitors.
It’s important to clarify that any modeling can only predict such interactions, but not prove their presence. The research cycle consists of at least three stages: modeling, synthesis of potentially active structures, and biological (pharmaceutical) testing of the necessary activity – real, rather than calculated effectiveness of the drug. Modeling alone, just like any other theoretical research, means nothing without following experimental confirmations. That’s why now is time for extensive practical work on validating the results received as part of ‘on-top docking’.
The tests that were performed on July 27, 2020, at Reaction Biology Corp., a certified laboratory in the U.S., demonstrated that disulfiram really inhibits Mpro in 100 nm concentration, which confirmed the results of the modeling. Unfortunately, the second substance – neratinib – demonstrated activity on Mpro, but it was insufficient for clinical use. On September 1, 2020, clinicians will start drug trials in vitro and in experimental treatments of patients with SARS-Cov-2.
Chinese biochemists carried out a massive experimental search for active structures simultaneously and independently of the Russian researchers. They have also detected potential activity of disulfiram to the SARS-CoV-2 virus main protease Mpro. Unfortunately, they did it two weeks earlier than the Russian chemists, so the publication in Nature is theirs (the paper will be issued in August). This serves as additional evidence of the importance of having powerful computational resources for modelling and capabilities for biological experiments.
“We need an opportunity to ‘claim’ the results immediately in a high-level Russian chemistry journal. And there are only a few of them. Unfortunately, if we approve only publications in 1st and 2nd quartile journals, which are exceptionally international, such Russian journals will never appear.” Igor Svitanko, Doctor of Sciences (Chemistry), Professor at the HSE Joint Department of Organic Chemistry with the RAS Zelinsky Institute of Organic Chemistry
Meanwhile, the main achievement is the demonstration that the ‘on-top docking’ approach is working and returns quite realistic and controllable results. The team’s plans for late 2020 and 2021 include molecular modeling of treatments for diseases that have demonstrated their harmfulness but have not yet spread over the world.
It’s important to mention that any molecular modeling requires significant computational resources, and before cooperating with HSE University, the chemists had been able to use their method only on very limited terms. Today, they have access to HSE University’s powerful supercomputer, which can help them search among existing drugs and perform targeted synthesis of new pharmaceutical products.
This is a brilliant example of fruitful cooperation between a university and a Russian Academy of Sciences institute. An obvious next step in such academic cooperation is organizing a Laboratory of Molecular Modelling at HSE University. This laboratory would not only create drugs, but it would model various chemical processes both by means of docking or other simple methods and by more universal and complicated quantum chemistry methods.
Meanwhile, the global chemistry community is facing the next challenge – modeling the structure of an inhibitor for the protein of the G4 EA H1N1 virus – a novel swine flu that was recently been detected in China. Researchers believe that this infection is much more dangerous and transfers more quickly from a human to human than COVID-19. To deal with it, researchers will need support, both in terms of resources and tools, and they will also need support organizing productive academic work and priority setting.
Reference: “Computational identification of disulfiram and neratinib as putative SARS-CoV-2 main protease inhibitors” by Victor S. Stroylova and Igor V.Svitanko, 4 August 2020, Mendeleev Communications.
DOI: 10.1016/j.mencom.2020.07.004
Never miss a breakthrough: Join the SciTechDaily newsletter.
Follow us on Google and Google News.