Results of large biobank study by Mount Sinai researchers may help doctors better assess true disease risk.
Imagine getting a positive result on a genetic test. The doctor tells you that you have a “pathogenic genetic variant,” or a DNA sequence that is known to raise the chances for getting a disease like breast cancer or diabetes. But what exactly are those chances — 10 percent? Fifty percent? One hundred? Currently, that is not an easy question to answer.
Large-Scale Biobank Study and Surprising Findings
To address this need, researchers at the Icahn School of Medicine at Mount Sinai analyzed the DNA sequences and electronic health record data of thousands of individuals stored in two massive biobanks. Overall, they discovered that the chance a pathogenic genetic variant may actually cause a disease is relatively low — about 7 percent. Nonetheless, they also found that some variants, such as those associated with breast cancer, are linked to a wide range of risks for disease. The results, published in JAMA, could alter the way the risks associated with these variants are reported, and one day, help guide the way physicians interpret genetic testing results.
“A major goal of this study was to produce helpful, advanced statistics which quantitatively assess the impact that known disease-causing genetic variants may have on an individual’s risk to disease,” said Ron Do, PhD, Associate Professor of Genetics and Genomic Sciences and a member of The Charles Bronfman Institute for Personalized Medicine at Icahn Mount Sinai.
Over the past 20 years scientists have discovered hundreds of thousands of variants that could cause a variety of diseases. However, due to the nature of these discoveries, it has been difficult to estimate — or provide statistics on — the true risk of this happening for each gene variant. So far, most estimates have been based on studies involving a small number of subjects, who were either part of a family that had a history of having a disease or were recruited at disease-specific clinics. But studies like these that do not use randomly chosen large populations may produce overestimates of the risk posed by variants.
Unveiling Disease Risks Through Biobank Data
In this study, the researchers tackled the issue by searching large-scale DNA sequencing data of 72,434 individuals for 37,780 known variants and then scanning each individual’s health records for a corresponding disease diagnosis. The extensive search involved 29,039 participants in Mount Sinai’s BioMe® Biobank program and 43,395 participants who were part of the UK Biobank.
The study was led by Iain S. Forrest, an MD-PhD candidate in Dr. Do’s lab who found inspiration from prior clinical experience he had as part of a postbaccalaureate fellowship at the National Institutes of Health (NIH).
“The idea for the study came out of a brainstorming session,” said Mr. Forrest. “Dr. Do and I discussed the need to have a better system for classifying disease risk. Currently, variants are categorized by broad labels such as ‘pathogenic’ or ‘benign.’ As I learned in the clinic, there’s a lot of grey area with these labels. That’s when we realized that the biobanks which link DNA sequence data to electronic health records are an unparalleled opportunity to address this need.”
Insights and Implications for Disease Risk Assessment
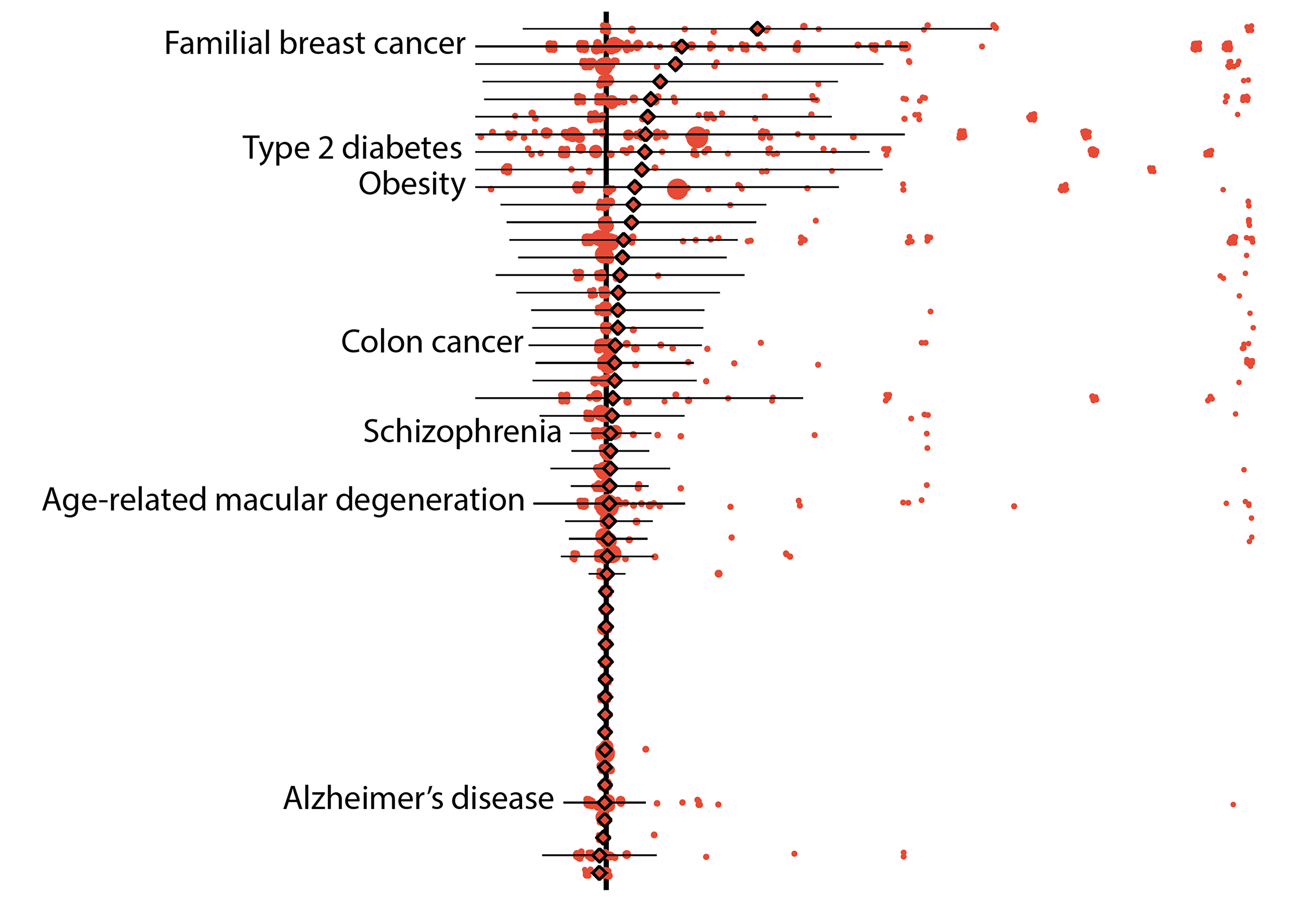
Initial results showed that 157 diseases in their data set could be linked to 5,360 variants that were defined as either “pathogenic” by ClinVar, a widely referenced, NIH-supported public library, or “loss-of-function” as predicted by bioinformatic algorithms. On average, the “penetrance,” or chance that a variant was linked to a disease diagnosis, was low, specifically 6.9 percent. Likewise, the average risk difference, which describes the increase in disease risk for an individual who has the variant over an individual who does not have it, was also low.
“At first I was quite surprised by the results. The risks we discovered were lower than I expected,” said Dr. Do. “These results raise questions about how we should be classifying the risks of these variants.”
Despite these results, the risks associated with some genetic variants remained high. For instance, pathogenic variants of the breast cancer genes BRCA1 and BRCA2 both averaged 38 percent penetrance, with individual variants falling between zero and 100 percent.
Further results demonstrated other advantages of using biobank data. In one example, the researchers were able to calculate the risks of individual variants that are associated with age-related disorders, such as some forms of type 2 diabetes and breast and prostate cancers. On average, the penetrance of these variants was about 10 percent for individuals over 70 years of age whereas it was about 8 percent for those who were older than 20.
The team also found that the presence of some variants could depend on an individual’s ethnicity and identified more than 100 variants that are specifically found in individuals of non-European descent.
Finally, the authors listed several potential ways the study itself could have under- or overestimated the risks reported.
“While more research is needed to be done, we feel that this study is a good first step towards eventually providing doctors and patients with the accurate and nuanced information they need to make more precise diagnoses,” said Dr. Do.
Reference: “Population-Based Penetrance of Deleterious Clinical Variants” by Iain S. Forrest, BS; Kumardeep Chaudhary, PhD; Ha My T. Vy, PhD; Ben O. Petrazzini, BS; Shantanu Bafna, MS; Daniel M. Jordan, PhD; Ghislain Rocheleau, PhD; Ruth J. F. Loos, PhD; Girish N. Nadkarni, MD; Judy H. Cho, MD and Ron Do, PhD, 25 January 2022, .
DOI: 10.1001/jama.2021.23686
This work was supported by the National Institutes of Health (GM124836, GM007280, HL139865, and HL155915).
Never miss a breakthrough: Join the SciTechDaily newsletter.
Follow us on Google and Google News.