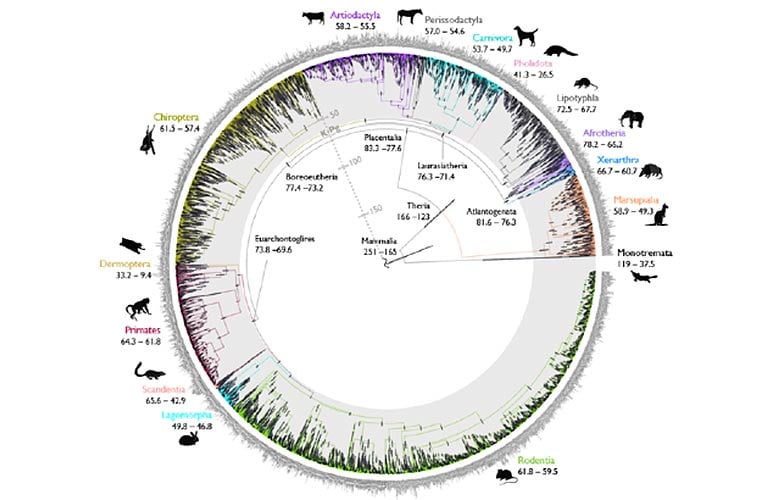
A new study, published on December 22, 2021, in the journal Nature, has provided the most detailed timeline of mammal evolution to date.
The research describes a new and fast computational approach to obtain precisely dated evolutionary trees, known as ‘timetrees’. The authors used the novel method to analyze a mammal genomic dataset and answer a long-standing question around whether modern placental mammal groups originated before or after the Cretaceous-Palaeogene (K-Pg) mass extinction, which wiped out over 70 percent of all species, including all dinosaurs.
The findings confirm the ancestors of modern placental mammal groups postdate the K-Pg extinction that occurred 66 million years ago, settling a controversy around the origins of modern mammals. Placental mammals are the most diverse group of living mammals, and include groups such as primates, rodents, cetaceans, carnivorans, chiropterans (bats) as well as humans.
The research team was led by Dr. Mario dos Reis (Queen Mary University of London) and Professor Phil Donoghue (University of Bristol), and included scientists from Queen Mary, University of Bristol, UCL, Imperial College London, and the University of Cambridge.
Dr. Sandra Álvarez-Carretero, lead author of the paper from UCL (then at Queen Mary), says: “By integrating complete genomes in the analysis and the necessary fossil information, we were able to reduce uncertainties and obtain a precise evolutionary timeline. Did modern mammal groups co-exist with the dinosaurs, or did they originate after the mass extinction? We now have a definite answer.”
“The timeline of mammal evolution is perhaps one of the most contentious topics in evolutionary biology. Early studies provided origination estimates for modern placental groups deep in the Cretaceous, in the dinosaur era. The past two decades had seen studies moving back and forth between post- and pre-K-Pg diversification scenarios. Our precise timeline settles the issue,” adds Prof Donoghue, co-senior author of the paper.
Fast Approach for Genome Analysis
With worldwide sequencing projects now producing hundreds to thousands of genome sequences, and with imminent plans to sequence more than a million species, evolutionary biologists will soon have a wealth of information at their hands. However, current methods to analyze the vast genomic datasets available and create evolutionary timelines are inefficient and computationally expensive.
“Inferring evolutionary timelines is a fundamental goal of biology. However, state-of-the-art methods rely on using computers to simulate evolutionary timelines and assess the most plausible ones. In our case, this was difficult due to the gigantic dataset analyzed, involving genetic data from almost 5,000 mammal species and 72 complete genomes,” Dr. dos Reis says.
In this study, the researchers developed a new, fast Bayesian approach to analyze large numbers of genome sequences, whilst also accounting for uncertainties within the data. “We solved the computational hurdles by dividing the analysis in sub-steps: first simulating timelines using the 72 genomes and then using the results to guide the simulations on the remaining species. Using genomes reduces uncertainty because it allows rejection of unplausible timelines from the simulations,” says Dr dos Reis.
“Our data processing pipeline sourced as much genomic data for as many mammal species as possible. This was challenging because genetic databases contain inaccuracies and we had to develop a strategy to identify poor quality samples or mislabelled data that had to be removed,” adds Dr. Asif Tamuri, co-lead author of the paper from UCL, who was responsible for assembling the mammal genomic dataset.
More Efficient and Sustainable
Using their novel approach, the team was able to reduce computation time for this complex analysis from decades to months. “If we had tried to analyze this large mammal dataset in a supercomputer without using the Bayesian method we have developed, we would have had to wait decades to infer the mammal timetree. Just imagine how long this analysis could take if we were to use our own PCs,” says Dr. Álvarez-Carretero. “In addition, we managed to reduce computation time by a factor of 100. This new approach not only allows the analysis of genomic datasets, but also, by being more efficient, substantially reduces the CO2 emissions released due to computing,” Dr. Álvarez-Carretero continues.
The method developed in the study could be used to tackle other contentious evolutionary timelines that require analysis of large datasets. By integrating the novel Bayesian approach with the forthcoming genomes from the Darwin Tree of Life and Earth BioGenome projects, the idea of estimating a reliable evolutionary timescale for the Tree of Life now seems within reach.
Reference: “A Species-Level Timeline of Mammal Evolution Integrating Phylogenomic Data” by Sandra Álvarez-Carretero, Asif U. Tamuri, Matteo Battini, Fabrícia F. Nascimento, Emily Carlisle, Robert J. Asher, Ziheng Yang, Philip C. J. Donoghue and Mario dos Reis, 22 December 2021, Nature.
DOI: 10.1038/s41586-021-04341-1
Never miss a breakthrough: Join the SciTechDaily newsletter.
Follow us on Google and Google News.
8 Comments
The summary seems somewhat too succinct, looking at the picture which is far more informative. Ancestors of most modern groups may postdate the K-Pg boundary, but their last common ancestor predates it, which means that those ‘groups’ already had separated and existed before -66 MY BP. There was one and only one primate, for instance, who survived the impact and has now-living descendants, but who was already distinct from the ancestor of carnivores, ungulates etc
The paper itself is fairly succinct since it integrates massive amounts of information, most of which is relegated to Methods and Supplement sections. This is also a methods paper, so the mammal result is a showcase – an apt one since several of its authos have been involved in earlier attempts. For instance, Dr. Mario dos Reis himself published a massive paper a few years ago that came to the opposite result on origins of modern mammals.
Speaking of austere, I believe Dwrin’s estimate of 1 in 1,000 species being fossilized and found may still be fairly accurate. So your stem primate may have been a massive stem clade. It is often harder to model diversity than clock times.
*authors*
In the second paragraph, I believe you meant predate rather than postdate.
Depends on what you mean by “ancestors”. The abstract is a more precise description of the tree:
“For example, we confidently reject an explosive model of placental mammal origination in the Paleogene8 and show crown Placentalia originated in the Late Cretaceous with unambiguous ordinal diversification in the Paleocene/Eocene.”
The press release may have been thinking of the “unambiguous … diversification”.
Considering the imprecision and incompleteness of the fossil record, is this just precision for it’s own sake?
No – the paper demonstrates how more data and better methods improve and further the science.
Some of this data is hard to swallow. Marsupials are characterized as having evolved well into the Paleocene, but unless they evolved from Multituberculates, that is essentially impossible. And a middle Cretaceous origin for Monotremes, which are two middle ears away from being Therapsids, is also completely implausible.