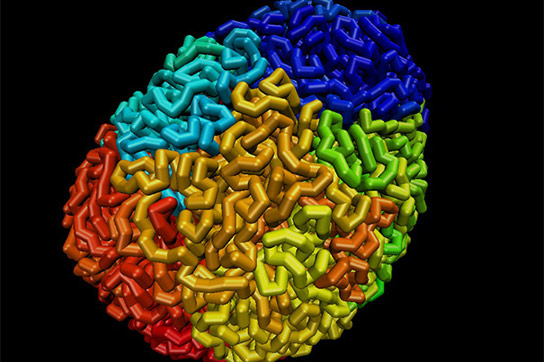
DNA forms loops and domains inside the nucleus. Credit: Adrian Sanborn, Erez Lieberman Aiden
A team of researchers has created the first 3D maps of entire folded genomes, identifying approximately 10,000 folding loops in the human genome.
In a triumph for cell biology, researchers have assembled the first high-resolution, 3-D maps of entire folded genomes and found a structural basis for gene regulation, a kind of “genomic origami” that allows the same genome to produce different types of cells. The research appears online Thursday in the journal Cell.
A central goal of the five-year project, a collaboration among researchers at Harvard University, Baylor College of Medicine, Rice University, and the Broad Institute of Harvard and MIT, was to identify the loops in the human genome. Loops form when two bits of DNA that are far apart in the genome sequence end up in close contact in the folded version of the genome in a cell’s nucleus.
Researchers used a technology called “in situ Hi-C” to collect billions of snippets of DNA that were later analyzed for signs of loops. The team found that loops and other genome folding patterns are an essential part of genetic regulation.
“More and more, we’re realizing that folding is regulation,” said study co-first author Suhas Rao, a researcher at Baylor’s Center for Genome Architecture and a 2012 graduate of Harvard College. “When you see genes turn on or off, what lies behind that is a change in folding. It’s a different way of thinking about how cells work.”
Co-first author Miriam Huntley, a doctoral student at the Harvard School of Engineering and Applied Sciences (SEAS), said, “Our maps of looping have revealed thousands of hidden switches that scientists didn’t know about before. In the case of genes that can cause cancer or other diseases, knowing where these switches are is vital.”
Senior author Erez Lieberman Aiden, Ph.D. ’10, formerly a junior fellow in the Harvard Society of Fellows, is now assistant professor of genetics at Baylor and of computer science and computational and applied mathematics at Rice. Aiden said the work began five years ago, shortly after he and colleagues at the Broad Institute published a groundbreaking study introducing the Hi-C methodology for sequencing genomes in 3-D.
“The 2009 study was a great proof of principle, but when we looked at the actual maps, we couldn’t see fine details,” Aiden said. “It took us a few years to get the resolution to a biologically usable level. The new maps allow us to really see, for the first time, what folding looks like at the level of individual genes.”
The work to refine Hi-C and produce full-genome maps with gene-level resolution continued when Aiden moved to Houston in 2013, established the Center for Genome Architecture at Baylor and joined the Center for Theoretical Biological Physics at Rice. Aiden, who earned his master’s degree in applied physics and Ph.D. in applied mathematics at SEAS, credited Huntley and Rao with leading the research effort.
In addition to the challenge of overhauling the Hi-C experimental design, the team faced significant computational hurdles.
“In 2009, we were dividing the genome into one-million-base blocks, and here we are dividing it into 1,000-base blocks,” said Huntley, who is Aiden’s student. “Since any block can collide with any other block, we end up with a problem that is a million-fold more complicated. The overall database is simply vast.”
Identifying the loops was yet another challenge.
“Ordinary computer CPUs [central processing units] are not well-adapted for the task of loop detection,” Rao said. “To find the loops, we had to use GPUs, processors that are typically used for producing computer graphics.”
Huntley said new methods were also developed to speed the data processing and reduce experimental “noise,” irregular fluctuations that tend to obscure weak signals in the data.
“We faced a real challenge because we were asking: ‘How do each of the millions of pieces of DNA in the database interact with each of the other millions of pieces?’” Huntley said. “Most of the tools that we used for this paper we had to create from scratch because the scale at which these experiments are performed is so unusual.”
The big-data tools created for the study included parallelized pipelines for high-performance computer clusters, dynamic programming algorithms, and custom data structures. Rao said the group also relied heavily on data-visualization tools created by co-authors Neva Durand and James Robinson.
“When studying big data, there can be a tendency to try to solve problems by relying purely on statistical analyses to see what comes out, but our group has a different mentality,” Rao said. “Even though there was so much data, we still wanted to be able to look at it, visualize it, and make sense of it. I would say that almost every phenomenon we observed was first seen with the naked eye.”
Through these methods, the team discovered a series of rules about how and where loops can form in the genome.
“If DNA were a shoestring, you could make a loop anywhere. But within the cell, the formation of loops is highly constrained,” said Rao. “The loops we see almost all span fewer than 2 million genetic letters; they rarely overlap; and they are almost always associated with a single protein, called CTCF.” CTCF is involved in regulation of the 3-D structure of chromatin, the building block of chromosomes.
“The most stunning discovery was about how CTCF proteins form a loop” said Eric Lander, a corresponding author on the paper. “Even when they are far apart, the CTCF elements that form a loop must be pointing at each other — forming a genomic yin and yang.” Lander is director of the Broad Institute, professor of biology at Massachusetts Institute of Technology, and professor of systems biology at Harvard Medical School.
Interestingly, the team found that the largest loops in the genome are present only in women. Huntley pointed out that “the copy of the X chromosome that is off in females contains gigantic loops that are up to 30 times the size of anything we see in males.”
The researchers also found that many of the loops present in humans are also present in mice, implying that these specific folds have been preserved over nearly a hundred million years of evolution.
“Our findings suggest that mammals share not only similar 1-D genome sequences, but also similar 3-D genome-folding patterns,” said Aiden.
Reference: “A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping” by Suhas S.P. Rao, Miriam H. Huntley, Neva C. Durand, Elena K. Stamenova, Ivan D. Bochkov, James T. Robinson, Adrian L. Sanborn, Ido Machol, Arina D. Omer, Eric S. Lander and Erez Lieberman Aiden, 11 December 2014, Cell.
DOI: 10.1016/j.cell.2014.11.021
Additional co-authors include Elena Stamenova, Ivan Bochkov, Adrian Sanborn, Ido Machol, and Arina Omer at Baylor. The research was supported by the National Science Foundation, the National Institutes of Health, the National Human Genome Research Institute, NVIDIA, IBM, Google, the Cancer Prevention and Research Institute of Texas, and the McNair Medical Institute.




Be the first to comment on "Scientists Create the First 3D Maps of Entire Folded Genomes"