STAMPScreen Pipeline Helps Streamline Genetic Studies in Mammalian Cells
Today’s genetic engineers have a plethora of resources at their disposal: an ever-increasing number of massive datasets available online, highly precise gene editing tools like CRISPR, and cheap gene sequencing methods. But the proliferation of new technologies has not come with a clear roadmap to help researchers figure out which genes to target, which tools to use, and how to interpret their results. So, a team of scientists and engineers at Harvard’s Wyss Institute for Biologically Inspired Engineering, Harvard Medical School (HMS), and the MIT Media Lab decided to make one.
The Wyss team has created an integrated pipeline for performing genetic screening studies, encompassing every step of the process from identifying target genes of interest to cloning and screening them quickly and efficiently. The protocol, called Sequencing-based Target Ascertainment and Modular Perturbation Screening (STAMPScreen), is described in Cell Reports Methods, and the associated open-source algorithms are available on GitHub.

“STAMPScreen is a streamlined workflow that makes it easy for researchers to identify genes of interest and perform genetic screens without having to guess which tool to use or what experiments to perform to get the results they want,” said corresponding author Pranam Chatterjee, Ph.D., a former graduate student at the MIT Media Lab who is now the Carlos M. Varsavsky Research Fellow at HMS and the Wyss Institute. “It is fully compatible with many existing databases and systems, and we hope that many scientists are able to take advantage of STAMPScreen to save themselves time and improve the quality of their results.”
Frustration Is the Mother of Invention
Chatterjee and Christian Kramme, a co-first author of the paper, were frustrated. The two scientists were trying to explore the genetic underpinnings of different aspects of biology — like fertility, aging, and immunity — by combining the strengths of digital methods (think algorithms) and genetic engineering (think gene sequencing). But they kept running into problems with the various tools and protocols they were using, which are commonplace in science labs.
The algorithms that purported to sift through an organism’s genes to identify those with a significant impact on a given biological process could tell when a gene’s expression pattern changed, but didn’t provide any insight into the cause of that change. When they wanted to test a list of candidate genes in living cells, it wasn’t immediately clear what type of experiment they should run. And many of the tools available to insert genes into cells and screen them were expensive, time-consuming, and inflexible.

“I was using methods known as Golden Gate and Gateway to clone genes into vectors for screening experiments, and it took me months and thousands of dollars to clone 50 genes. And using Gateway, I couldn’t physically barcode the genes to identify which one got into which vector, which was a crucial requirement for my downstream sequencing-based experimental design. We figured there had to be a better way to do this kind of research, and when we couldn’t find one, we took on the challenge of creating it ourselves,” said Kramme, who is a graduate student at the Wyss Institute and HMS,
Kramme teamed up with co-first author and fellow Church lab member Alexandru Plesa, who was experiencing identical frustrations making gene vectors for his project. Kramme, Plesa, and Chatterjee then set to work outlining what would be required to make an end-to-end platform for genetic screening that would work for all of their projects, which ranged from protein engineering to fertility and aging.
From Bits to the Bench
To improve the earliest stage of genetic research — identifying genes of interest to study — the team created two new algorithms to help meet the need for computational tools that can analyze and extract information from the increasingly large datasets that are being generated via next-generation sequencing (NGS). The first algorithm takes the standard data about a gene’s expression level and combines it with information about the state of the cell, as well as information about which proteins are known to interact with the gene. The algorithm gives a high score to genes that are highly connected to other genes and whose activity is correlated with large, cell-level changes. The second algorithm provides more high-level insight by generating networks to represent the dynamic changes in gene expression during cell-type differentiation and then applying centrality measures, such as Google’s PageRank algorithm, to rank the key regulators of the process.
“The computational part of genetic studies is like a Jenga game: if each block in the tower represents a gene, we’re looking for the genes that make up the base of the Jenga tower, the ones that hold the whole thing up. Most algorithms can only tell you which genes are in the same row as each other, but ours allow you to home in on how far up or down the tower they are, so you can quickly identify the ones that have the biggest influence on the cell state in question,” said Chatterjee.
Once the target genes have been identified, the STAMPScreen protocol moves from the laptop to the lab, where experiments are performed to disrupt those genes in cells and see what effect that perturbation has on the cell. The team of researchers systematically evaluated multiple gene perturbation tools including complementary DNA (cDNA) and several versions of CRISPR in human induced pluripotent stem cells (hiPSCs), the first known head-to-head comparisons performed entirely in this highly versatile yet challenging cell type.
They then created a new tool that allows CRISPR and cDNA to be used within the same cell to unlock synergies between the two methods. For example, CRISPR can be used to turn off expression of all isoforms of a gene, and cDNA can be used to sequentially express each isoform individually, allowing more nuanced genetic studies and greatly reducing background expression of off-target genes.
Scanning Library Barcodes
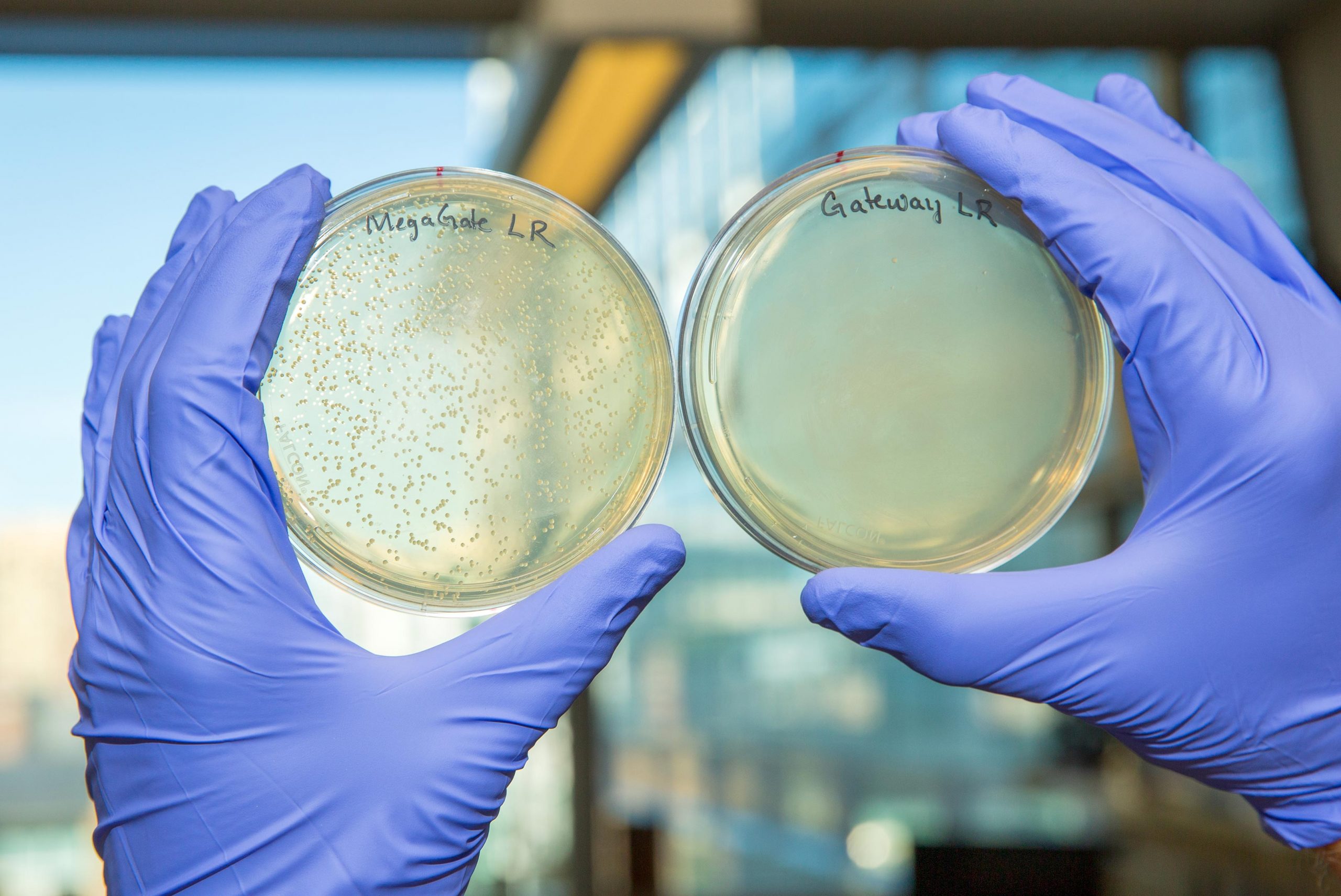
The next step in many genetic experiments is generating a screening library for introducing genes into cells and observing their effects. Typically, gene fragments are inserted into bacterial plasmids (circular pieces of DNA) using methods that work well for small pieces of DNA, but are cumbersome to use when inserting larger genes. Many of the existing methods also rely on a technique called Gateway, which uses a process called lambda phage recombination and the production of a toxin to kill off any bacteria that did not receive a plasmid with the gene of interest. The toxin in these plasmids is often cumbersome to work with in the lab, and can be inadvertently inactivated when a “barcode” sequence is added to a vector to help researchers identify which gene-bearing plasmid the vector received.
Kramme and Plesa were working with Gateway when they realized that these problems could be solved if they eliminated the toxin and replaced it with short sequences on the plasmid that would be recognized and cut by a type of enzyme called meganucleases. Meganuclease recognition sequences do not appear in the genes of any known organism, thus ensuring that the enzyme will not accidentally cut the inserted gene itself during cloning. These recognition sequences are naturally lost when a plasmid receives a gene of interest, making those plasmids immune to meganuclease. Any plasmids that do not successfully receive the gene of interest, however, retain these recognition sequences and are cut to pieces when a meganuclease is added, leaving only a pure pool of plasmids containing the inserted gene. The new method, which the researchers dubbed MegaGate, had a cloning success rate of 99.8% and also allowed them to barcode their vectors with ease.
“MegaGate not only solves many of the problems that we kept running into with older cloning methods, it is also compatible with many existing gene libraries like the TFome and hORFeome. You can essentially take Gateway and meganucleases off the shelf, put them together with a library of genes and a library of barcoded destination vectors, and two hours later you have your barcoded genes of interest. We’ve cloned nearly 1,500 genes with it, and have yet to have a failure,” said Plesa, who is a graduate student at the Wyss Institute and HMS.
Finally, the researchers demonstrated that their barcoded vectors could be successfully inserted into living hiPSCs, and pools of cells could be analyzed using NGS to determine which delivered genes were being expressed by the pool. They also successfully used a variety of methods, including RNA-Seq, TAR-Seq, and Barcode-Seq, to read both the genetic barcodes and the entire transcriptomes of hiPSCs, enabling researchers to use whichever tool they are most familiar with.
The team anticipates that STAMPScreen could prove useful for a wide variety of studies, including pathway and gene regulatory network studies, differentiation factor screening, drug and complex pathway characterizations, and mutation modeling. STAMPScreen is also modular, allowing scientists to integrate different parts of it into their own workflows.
“There’s a treasure trove of information housed in publicly available genetic datasets, but that information will only be understood if we use the right tools and methods to analyze it. STAMPScreen will help researchers get to eureka moments faster and speed up the pace of innovation in genetic engineering,” said senior author George Church, Ph.D., a Wyss Core Faculty member who is also a Professor of Genetics at HMS and Professor of Health Sciences and Technology at Harvard and MIT.
“At the Wyss Institute we aim for impactful ‘moonshot’ solutions to pressing problems, but we know that to get to the moon, we have to first build a rocket. This project is a great example of how our community innovates on-the-fly to enable scientific breakthroughs that will change the world for the better,” said Wyss Founding Director Don Ingber, M.D., Ph.D., who is also the Judah Folkman Professor of Vascular Biology at HMS and the Vascular Biology Program at Boston Children’s Hospital, as well as Professor of Bioengineering at Harvard John A. Paulson School of Engineering and Applied Sciences.
Reference: “An Integrated Pipeline for Mammalian Genetic Screening” by Christian Kramme, Alexandru M. Plesa, Helen H. Wang, Bennett Wolf, Merrick Pierson Smela, Xiaoge Guo, Richie E. Kohman, Pranam Chatterjee and George M. Church, 27 September 2021, Cell Reports Methods.
DOI: 10.1016/j.crmeth.2021.100082
Additional authors of the paper include Helen Wang, Bennett Wolf, Merrick Smela, Xiaoge Guo, Ph.D., and Richie Kohman, Ph.D. from the Wyss Institute and HMS.
Never miss a breakthrough: Join the SciTechDaily newsletter.
Follow us on Google and Google News.