
In a major advance, scientists have assembled genomic sequences of 47 people from diverse backgrounds to create a pangenome, which offers a more accurate representation of human genetic diversity than the existing reference genome. This new pangenome will help researchers refine their understanding of the link between genes and diseases, and could ultimately help address health disparities.
For more than 20 years, scientists have relied on the human reference genome, a consensus genetic sequence, as a standard against which to compare other genetic data. Used in countless studies, the reference genome has made it possible to identify genes implicated in specific diseases and trace the evolution of human traits, among other things.
But it has always been a flawed tool. One of its biggest problems is that about 70 percent of its data came from a single man of predominantly African-European background whose DNA was sequenced during the Human Genome Project, the first effort to capture all of a person’s DNA. As a result, it can tell us little about the 0.2 to one percent of genetic sequence that makes each of the seven billion people on this planet different from each other, creating an inherent bias in biomedical data believed to be responsible for some of the health disparities affecting patients today. Many genetic variants found in non-European populations, for instance, aren’t represented in the reference genome at all.
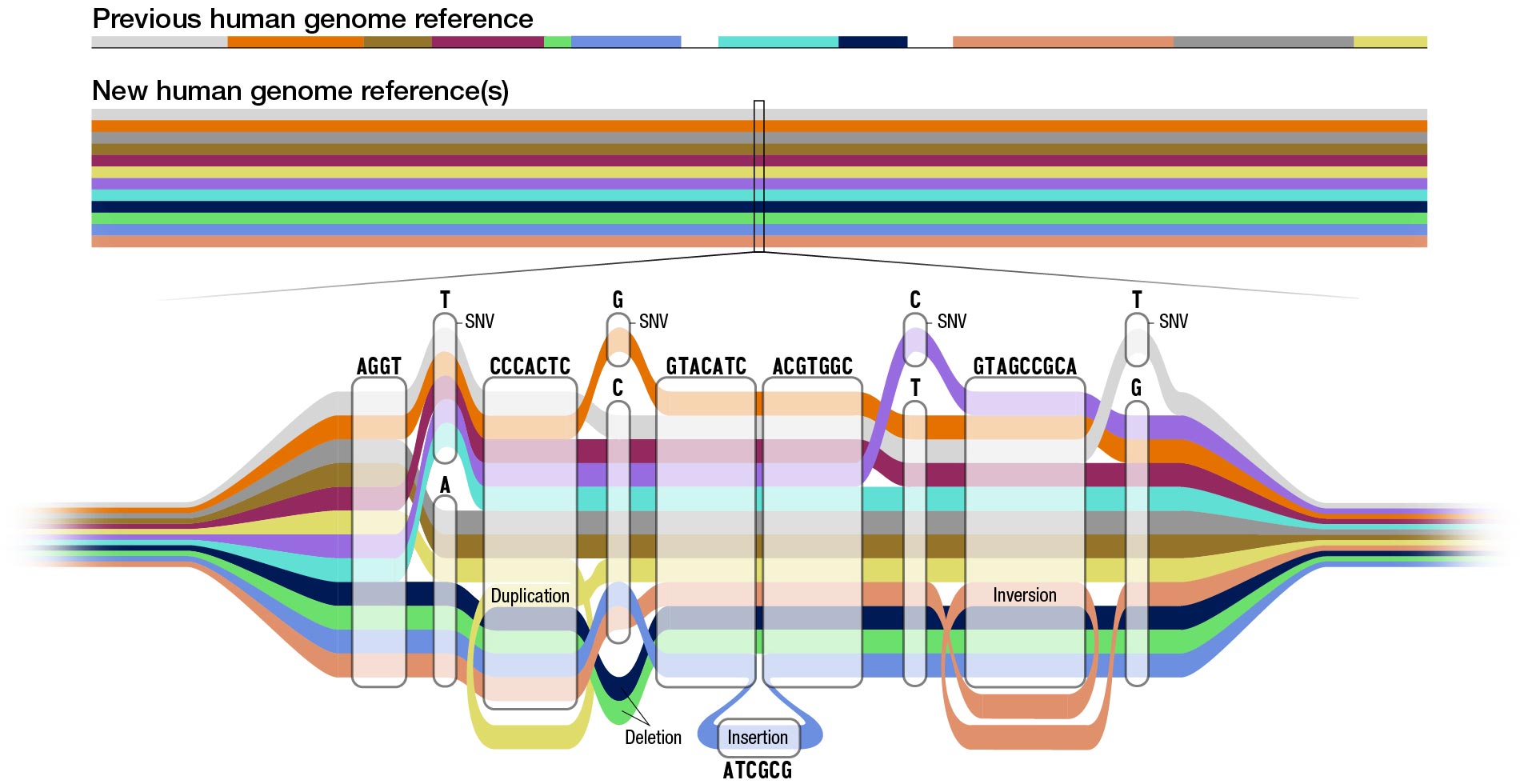
For years, researchers have called for a resource more inclusive of human diversity with which to diagnose diseases and guide medical treatments. Now scientists with the Human Pangenome Reference Consortium have made groundbreaking progress in characterizing the fraction of human DNA that varies between individuals. As they recently published in Nature, they’ve assembled genomic sequences of 47 people from around the world into a so-called pangenome in which more than 99 percent of each sequence is rendered with high accuracy.
Layered upon each other, these sequences revealed nearly 120 million DNA base pairs that were previously unseen.
While it’s still a work in progress, the pangenome is public and can be used by scientists around the world as a new standard human genome reference, says The Rockefeller University’s Erich D. Jarvis, one of the primary investigators.
“This complex genomic collection represents significantly more accurate human genetic diversity than has ever been captured before,” he says. “With a greater breadth and depth of genetic data at their disposal, and greater quality of genome assemblies, researchers can refine their understanding of the link between genes and disease traits, and accelerate clinical research.”
Sourcing Diversity
Completed in 2003, the first draft of the human genome was relatively imprecise, but it became sharper over the years thanks to filled-in gaps, corrected errors, and advancing sequencing technology. Another milestone was reached last year, when the final eight percent of the genome—mainly tightly coiled DNA that doesn’t code for protein and repetitive DNA regions—was finally sequenced.
Despite this progress, the reference genome remained imperfect, especially with respect to the critical 0.2 to one percent of DNA representing diversity. The Human Pangenome Reference Consortium (HPRC), a government-funded collaboration between more than a dozen research institutions in the United States and Europe, was launched in 2019 to address this problem.
At the time, Jarvis, one of the consortium’s leaders, was honing advanced sequencing and computational methods through the Vertebrate Genomes Project, which aims to sequence all 70,000 vertebrate species. His and other collaborating labs decided to apply these advances for high-quality diploid genome assemblies to revealing the variation within a single vertebrate: Homo sapiens.
To collect a diversity of samples, the researchers turned to the 1000 Genomes Project, a public database of sequenced human genomes that includes more than 2500 individuals representing 26 geographically and ethnically varied populations. Most of the samples come from Africa, home to the planet’s largest human diversity.
“In many other large human genome diversity projects, the scientists selected mostly European samples,” Jarvis says. “We made a purposeful effort to do the opposite. We were trying to counteract the biases of the past.”
It’s likely that gene variants that could inform our knowledge of both common and rare diseases can be found among these populations.
Mom, Dad, and Child
But to broaden the gene pool, the researchers had to create crisper, clearer sequences of each individual–and the approaches developed by members of the Vertebrate Genome Project and associated consortiums were used to solve a longstanding technical problem in the field.
Every person inherits one genome from each parent, which is how we end up with two copies of every chromosome, giving us what’s known as a diploid genome. And when a person’s genome is sequenced, teasing apart parental DNA can be challenging. Older techniques and algorithms have routinely made errors when merging parental genetic data for an individual, resulting in a cloudy view. “The differences between mom’s and dad’s chromosomes are bigger than most people realize,” Jarvis says. “Mom may have 20 copies of a gene and dad only two.”
With so many genomes represented in a pangenome, that cloudiness threatened to develop into a thunderstorm of confusion. So the HPRC homed in a method developed by Adam Phillippy and Sergey Koren at the National Institutes of Health on parent-child “trios”—a mother, a father, and a child whose genomes had all been sequenced. Using the data from mom and dad, they were able to clear up the lines of inheritance and arrive at a higher-quality sequence for the child, which they then used for pangenome analysis.
New Variations
The researchers’ analysis of 47 people yielded 94 distinct genome sequences, two for each set of chromosomes, plus the sex Y chromosome in males.
They then used advanced computational techniques to align and layer the 94 sequences. Of the 120 million DNA base pairs that were previously unseen or in a different location than they were noted to be in the previous reference, about 90 million derive from structural variations, which are differences in people’s DNA that arise when chunks of chromosomes are rearranged—moved, deleted, inverted, or with extra copies from duplications.
It’s an important discovery, Jarvis notes, because studies in recent years have established that structural variants play a major role in human health, as well as in population-specific diversity. “They can have dramatic effects on trait differences, disease, and gene function,” he says. “With so many new ones identified, there’s going to be a lot of new discoveries that weren’t possible before.”
Filling Gaps
The pangenome assembly also fills in gaps that were due to repetitive sequences or duplicated genes. One example is the major histocompatibility complex (MHC), a cluster of genes that code proteins on the surface of cells that help the immune system recognize antigens, such as those from the SARS-CoV-2 virus.
“They’re really important, but it was impossible to study MHC diversity using the older sequencing methods,” Jarvis says. “We’re seeing much greater diversity than we expected. This new information will help us understand how immune responses against specific pathogens vary among people.” It could also lead to better methods to match organ transplant donors with and patients, or identify people at risk for developing autoimmune disease.
The team has also uncovered surprising new characteristics of centromeres, which lie at the cruxes of chromosomes and conduct cell division, pulling apart as cells duplicate. Mutations in centromeres can lead to cancers and other diseases.
Despite having highly repetitive DNA sequences, “centromeres are so diverse from one haplotype to another, that they can account for more than 50 percent of the genetic differences between people or maternal and paternal haplotypes even within one individual,” Jarvis says. “The centromeres seem to be one of the most rapidly evolving parts of the chromosome.”
Relationship building
The current 47-people pangenome is just a starting point, however. The HPRC’s ultimate goal is to produce high-quality, nearly error-free genomes from at least 350 individuals from diverse populations by mid-2024, a milestone that would make it possible to capture rare alleles that confer important adaptive traits. Tibetans, for example, have alleles related to oxygen use and UV light exposure that enable them to live at high altitudes.
A major challenge in collecting this data will be to gain trust from communities that have seen past abuses of biological data; for example, there are no samples in the current study from Native American nor Aboriginal peoples, who have long been disregarded or exploited by scientific studies. But you don’t have to go far back in time to find examples of unethical use of genetic data: Just a few years ago, DNA samples from thousands of Africans in multiple countries were commercialized without the donors’ knowledge, consent, or benefit.
These offenses have sown mistrust against scientists among many populations. But by not being included, some of these groups could remain genetically obscure, leading to a perpetuation of the biases in the data—and to continued disparities in health outcomes.
“It’s a complex situation that’s going to require a lot of relationship building,” Jarvis says. “There’s greater sensitivity now.”
And even today, many groups are willing to participate. “There are individuals, institutions, and governmental bodies from different countries who are saying, ‘We want to be part of this. We want our population to be represented,’” Jarvis says. “We’re already making progress.”
For more on this breakthrough, see Human Pangenome Reference: A Deeper Understanding of Worldwide Genomic Diversity.
References:
“A draft human pangenome reference” by Wen-Wei Liao, Mobin Asri, Jana Ebler, Daniel Doerr, Marina Haukness, Glenn Hickey, Shuangjia Lu, Julian K. Lucas, Jean Monlong, Haley J. Abel, Silvia Buonaiuto, Xian H. Chang, Haoyu Cheng, Justin Chu, Vincenza Colonna, Jordan M. Eizenga, Xiaowen Feng, Christian Fischer, Robert S. Fulton, Shilpa Garg, Cristian Groza, Andrea Guarracino, William T. Harvey, Simon Heumos, Kerstin Howe, Miten Jain, Tsung-Yu Lu, Charles Markello, Fergal J. Martin, Matthew W. Mitchell, Katherine M. Munson, Moses Njagi Mwaniki, Adam M. Novak, Hugh E. Olsen, Trevor Pesout, David Porubsky, Pjotr Prins, Jonas A. Sibbesen, Jouni Sirén, Chad Tomlinson, Flavia Villani, Mitchell R. Vollger, Lucinda L. Antonacci-Fulton, Gunjan Baid, Carl A. Baker, Anastasiya Belyaeva, Konstantinos Billis, Andrew Carroll, Pi-Chuan Chang, Sarah Cody, Daniel E. Cook, Robert M. Cook-Deegan, Omar E. Cornejo, Mark Diekhans, Peter Ebert, Susan Fairley, Olivier Fedrigo, Adam L. Felsenfeld, Giulio Formenti, Adam Frankish, Yan Gao, Nanibaa’ A. Garrison, Carlos Garcia Giron, Richard E. Green, Leanne Haggerty, Kendra Hoekzema, Thibaut Hourlier, Hanlee P. Ji, Eimear E. Kenny, Barbara A. Koenig, Alexey Kolesnikov, Jan O. Korbel, Jennifer Kordosky, Sergey Koren, HoJoon Lee, Alexandra P. Lewis, Hugo Magalhães, Santiago Marco-Sola, Pierre Marijon, Ann McCartney, Jennifer McDaniel, Jacquelyn Mountcastle, Maria Nattestad, Sergey Nurk, Nathan D. Olson, Alice B. Popejoy, Daniela Puiu, Mikko Rautiainen, Allison A. Regier, Arang Rhie, Samuel Sacco, Ashley D. Sanders, Valerie A. Schneider, Baergen I. Schultz, Kishwar Shafin, Michael W. Smith, Heidi J. Sofia, Ahmad N. Abou Tayoun, Françoise Thibaud-Nissen, Francesca Floriana Tricomi, Justin Wagner, Brian Walenz, Jonathan M. D. Wood, Aleksey V. Zimin, Guillaume Bourque, Mark J. P. Chaisson, Paul Flicek, Adam M. Phillippy, Justin M. Zook, Evan E. Eichler, David Haussler, Ting Wang, Erich D. Jarvis, Karen H. Miga, Erik Garrison, Tobias Marschall, Ira M. Hall, Heng Li and Benedict Paten, 10 May 2023, Nature.
DOI: 10.1038/s41586-023-05896-x
“Increased mutation rate and gene conversion within human segmental duplications” by Mitchell R. Vollger, Philip C. Dishuck, William T. Harvey, William S. DeWitt, Xavi Guitart, Michael E. Goldberg, Allison N. Rozanski, Julian Lucas, Mobin Asri, Human Pangenome Reference Consortium, Katherine M. Munson, Alexandra P. Lewis, Kendra Hoekzema, Glennis A. Logsdon, David Porubsky, Benedict Paten, Kelley Harris, PingHsun Hsieh and Evan E. Eichler, 10 May 2023. Nature.
DOI: 10.1038/s41586-023-05895-y
Never miss a breakthrough: Join the SciTechDaily newsletter.
Follow us on Google and Google News.
4 Comments
To me, this smacks of intelligent design. Not mindless evolution.
Yes.
People intelligently designed a genetic survey covering most general ‘types’ of human being. Which, they say, might be a useful medical tool. Y’know, for use in overcoming all those plagues god sent to teach us to bow and scrape.
Sooner or later, in an infinite universe of events, two or more elements will combine or collide in such a way that they create a fusion of elements that in turn gather additional elements and by and by the whole becomes greater than the sum of its parts. That is not mindless, it is the law of percentages.
This happens a lot quicker when (a) the atoms are polar, i.e. have a positive and negative ‘end’, and (b) quantum mechanics blurs the line around just exactly what is where, when.