Findings reveal potential new strategy for treating people diagnosed with schizophrenia.
Columbia researchers have restored normal working memory to a mouse model of schizophrenia, eliminating a core symptom of the disorder that, in people, has proven virtually impossible to treat.
Working memory is a fundamental brain process used to retain and recall information on the fly, such as remembering a new phone number long enough to dial it. It is severely impaired in people with schizophrenia altering their reasoning, perception, and decision-making. By repurposing a drug currently in development for leukemia, the Columbia team repaired the mouse models’ dysfunctional brain cells, bringing those cells — and the animals’ working memory — back to a full and healthy state.
These findings, published today (October 9, 2019) in Neuron, challenge the generally accepted belief that cellular disruptions underlying memory issues in schizophrenia cannot be repaired once symptoms arise. They also hold immense promise for treating the more than 21 million people worldwide already diagnosed with schizophrenia.
“Schizophrenia is thought to be a neurodevelopmental disorder that begins years before it can actually be diagnosed, making the disease’s underlying aspects extremely difficult to understand and treat,” said Joseph Gogos, MD, Ph.D., a principal investigator at Columbia’s Mortimer B. Zuckerman Mind Brain Behavior Institute and the paper’s senior author. “Today’s paper shows a promising way forward: A way to use knowledge from genetic studies to identify drugs that restore normal cognitive and cellular function in the adult brain after the onset of disease.”
Today’s paper shows a promising way forward: A way to use knowledge from genetic studies to identify drugs that restore normal cognitive and cellular function in the adult brain after the onset of disease. — Joseph Gogos, MD, Ph.D.
Schizophrenia’s most well-known symptoms — paranoia, auditory hallucinations, and delusions — can often be controlled with antipsychotic medication. But disruptions to working memory, an underlying problem that affects virtually all patients, have remained largely untreatable. This has spurred renewed efforts to target the disease’s underlying causes. Such memory problems can make it difficult to maintain relationships or a job, essentially cutting off people with schizophrenia from the world around them.
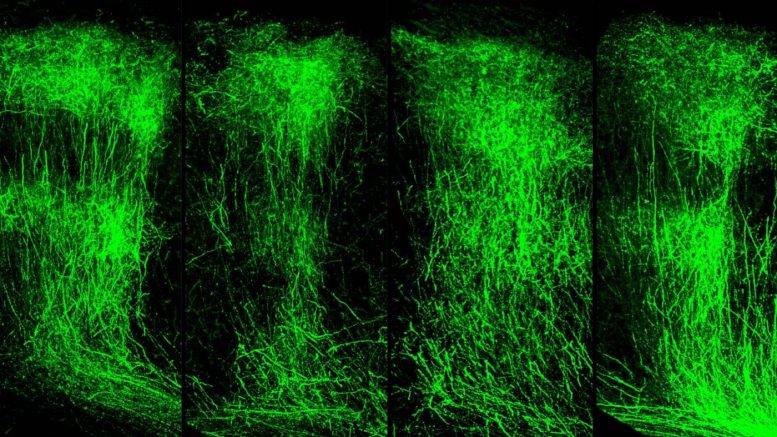
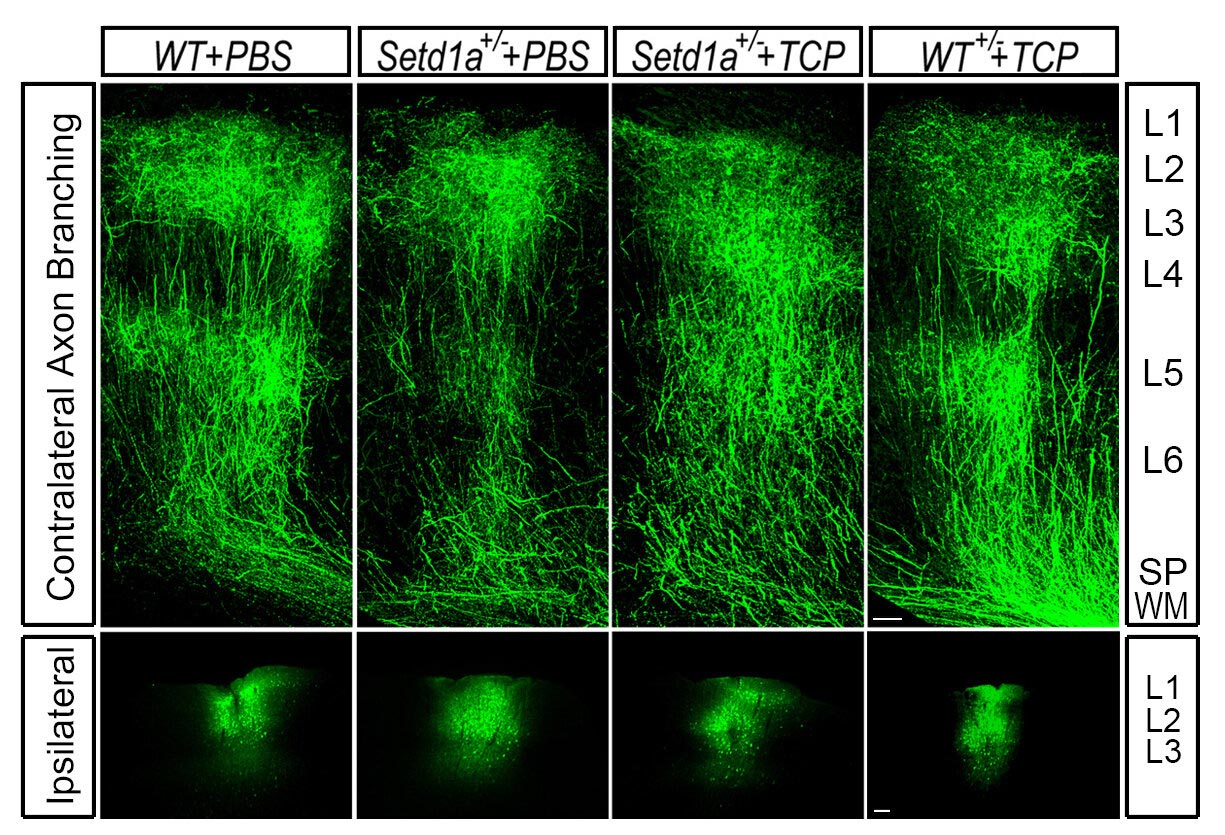
By administering an LSD1 inhibitor in adult SETD1A-deficient mice, the researchers rescued this stunted growth to near-normal levels. Credit: Jun Mukai/Gogos lab/Columbia’s Zuckerman Institute
For this study, the research team focused on the gene SETD1A, which makes a protein that can regulate, or change, the activity of other genes. Scientists have long known that this gene is important for embryos to grow properly. But in 2014, Dr. Gogos and his collaborators found that mutations in SETD1A are also associated with schizophrenia in people.
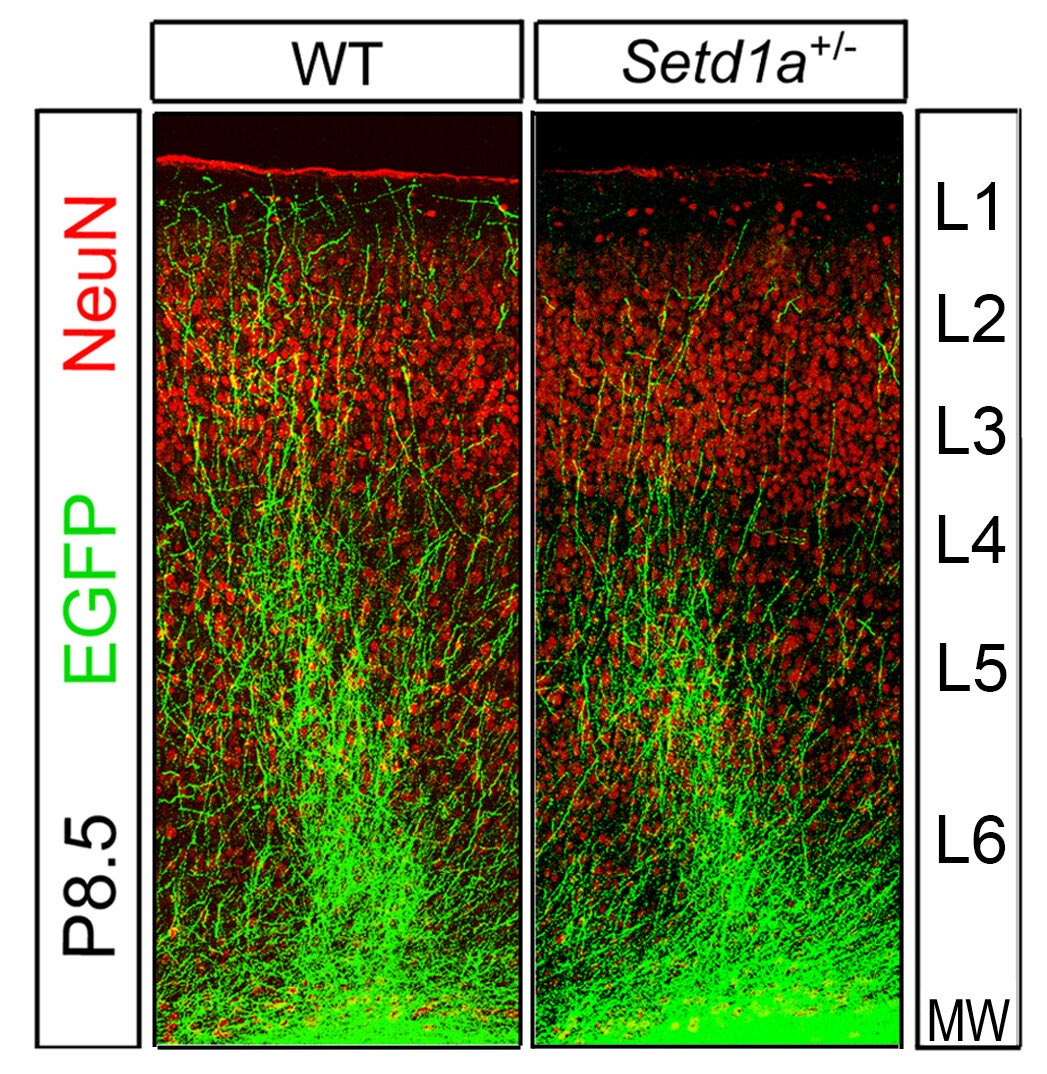
The researchers examined the behavior of mice with SETD1A genes that make half as much protein as usual, to mimic observations in patients. Compared to normal mice, these animals exhibited the telltale memory deficits associated with schizophrenia, and had difficulty navigating a simple maze.
Brain cells called neurons in the animals’ prefrontal cortex, a brain region critical for memory and complex behaviors, looked markedly different in these mice. Normally, neurons in this region have extendable branches. They use these branches to link up with other neurons and communicate with their cellular neighbors. But the neuronal branches of the SETD1A-deficient mice were short and stunted.
“The neurons’ misshapen axons prevented them from making the necessary connections to neurons next to them or in other parts of the brain,” said Jun Mukai, Ph.D., the paper’s co-first author who was an associate research scientist in the Gogos lab.
Searching for ways to fix the cells, the team began to think about ways to manipulate SETD1A. Dr. Gogos and his lab teamed up with Zuckerman Institute Principal Investigator and geneticist Stavros Lomvardas, Ph.D., and members of the Lomvardas lab, to tease apart SETD1A’s role in the brain.
“We found SETD1A to be a genomic multitasker,” said Enrico Cannavó, Ph.D., a postdoctoral research scientist in the Lomvardas lab and the paper’s co-first author. “Sometimes SETD1A turns a gene on, while other times it turns a gene off. This ability to dial up and down gene activity makes SETD1A complicated to study.”
Further complicating this work was the fact that there are no known means for manipulating SETD1A pharmacologically, such as with a drug, so the team identified a workaround. They found another gene called LSD1 (unrelated to the psychoactive substance LSD). When switched off, this gene nullified SETD1A’s harmful effects.
“Within a few weeks of administering an LSD1 inhibitor, the animals’ memory improved dramatically,” said Dr. Mukai. “Even more striking was what we observed in the animals’ brains: their axons grew in similar patterns to what we see in a healthy mouse brain.”
This observation demonstrated that the LSD1 inhibitor was not only acting on the memory deficits linked to schizophrenia but on the underlying molecular mechanisms that drive them.
“These results also elucidate a new role of SETD1A in the brain,” said Dr. Cannavó. “We’ve found definitive proof that not only does it guide early development, but it also supports ongoing functions in the adult brain, such as axonal growth.”
In fact, SETD1A likely impacts additional aspects of brain function. The team hypothesizes that SETD1A influences a host of other factors — such as other genes and proteins — and that the combined activity of all these factors ultimately cause the memory deficits observed in mutant mice.
Psychiatric disorders like schizophrenia have proven difficult to treat, in part because they lack a single cause, a single broken gene. With multiple genetic and environmental factors likely at play, these findings could pave the way to personalized medicines designed for individuals with SETD1A mutations — and perhaps even broader treatment strategies.
“Although SETD1A mutations exist in a small percentage of all schizophrenia patients, many people diagnosed with the disorder have issues similar to those caused by this mutation,” said Dr. Gogos, who is also a professor of physiology, cellular biophysics and neuroscience at Columbia’s Vagelos College of Physicians and Surgeons. “Thus, therapies that are specific to SETD1A may indeed have wider implications for schizophrenia as a whole.”
Moving forward, the team hopes to further elucidate the roles of SETD1A. They also hope to expand and fine-tune their investigations of LSD1 inhibitors as a therapeutic strategy.
“Several LSD1 inhibitors are in early-stage clinical trials for treating leukemia and other forms of cancer,” said Dr. Gogos. “We are exploring whether they could be repurposed to treat schizophrenia patients.”
###
Reference: “Recapitulation and Reversal of Schizophrenia-Related Phenotypes in Setd1a-Deficient Mice” by Jun Mukai, Enrico Cannavò, Gregg W. Crabtree, Ziyi Sun, Anastasia Diamantopoulou, Pratibha Thakur, Chia-Yuan Chang, Yifei Cai, Stavros Lomvardas, Atsushi Takata, Bin Xu and
Joseph A. Gogos, 9 October 2019, Neuron.
DOI: 10.1016/j.neuron.2019.09.014
This research was supported by the National Institutes of Health (R01MH080234, R01DA036894).
Never miss a breakthrough: Join the SciTechDaily newsletter.
Follow us on Google and Google News.
3 Comments
Really nice discovery, but we should remember that it’s only an animal model. Schizophrenia in mice is quite different than in human. We now know of over 200 genes associated with it in humans – and in this experiment, they just worked out how to tackle one murine one.
Great article, I have read a lot of articles on this topic, but I think your article has the most impressive and detailed content, I hope you will have more interesting songs. More to bring to the reader, thank you for sharing.
This could be BIG in Dementia, Chemo Brain Fog and Fibromyalgia Research!