Researchers from Yale University have made a fundamental discovery about EGFR signaling that may open the potential for new types of cancer drugs.
Many approved cancer therapies target a protein called epidermal growth factor receptor (EGFR) that regulates many crucial cellular processes and can speed the proliferation of tumor cells.
The EGFR protein spans the cell membrane, with one part outside the cell and one part inside. “When signaling molecules known as growth factors bind to the outside part of EGFR, they trigger the enzyme within the inside part, and initiate proliferation or other cellular responses,” said corresponding author Mark Lemmon, Ph.D., F.R.S., co-director of the Cancer Biology Institute at Yale Cancer Center and a professor of pharmacology at Yale School of Medicine.
“Researchers have long known that growth factors activate EGFR by “stitching” two receptor molecules together,” added Lemmon. “This paradigm has always suggested that the receptor has to be either “off” or “on”, so all EGFR drugs have been designed to shut off the receptor and thus shut off proliferation.”
However, there’s also been a longstanding puzzle in this field: The EGFR is regulated by a total of seven growth factors, which can make the cell take different actions. So, how can those different actions be driven by a single binding (and activation) scenario?
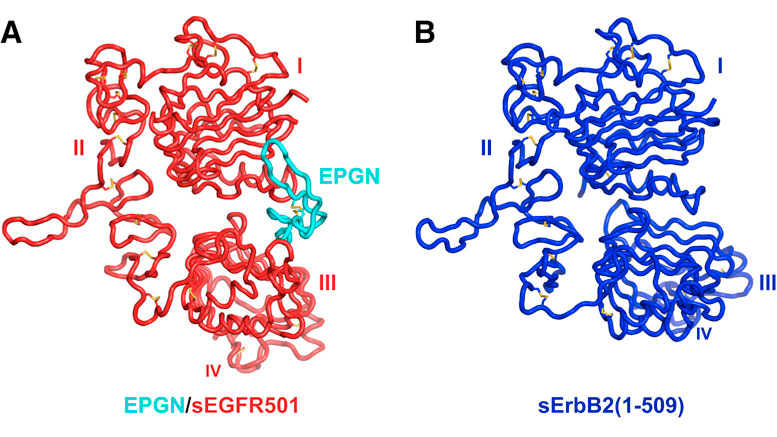
Lemmon and his colleagues began their research with X-ray crystallography—crystallizing the outer parts of the EGFR protein, shining X-rays on the crystals, collecting data on how the X-rays diffract, and then deriving the protein’s structure from those data. They then linked their structural results with biological mechanisms in cellular experiments.
Surprisingly, the researchers found that EGFR signaling is not just an on/off process controlled by stitching two receptors together. Instead, the growth factors can turn on the receptor in a spectrum of different ways, depending on the strength of the stitch and the timing of this binding.
“We can imagine that instead of therapeutics that just shut off EGFR, we might design new ones that encourage it to give a beneficial signal,” Lemmon said. For instance, his team showed that growth factors that stitch two receptors together only weakly – and for a short duration – signal the cell to begin to transform into another cell type rather than to proliferate. A biologic drug that mimics the actions of such growth factors might discourage cancer cells from proliferating, Lemmon speculated.
“There are cancer drugs against EGFR that really work, but we need new ways to develop them because cancers can develop resistance to all of them,” he pointed out.
The spectrum of effects from different EGFR binding mechanisms also might help to shed light on other biological mysteries such as the causes of liver cancer, where pathways that work in similar ways to EGFR signaling play major roles that have not been well explained, Lemmon added.
Co-lead authors on the paper were Daniel Freed of Yale and Nicholas Bessman of the University of Pennsylvania (now at Cornell Weill Medical College). Lead funding came from the National Institutes of Health.
Reference: “EGFR Ligands Differentially Stabilize Receptor Dimers to Specify Signaling Kinetics” by Daniel M. Freed, Nicholas J. Bessman, Anatoly Kiyatkin, Emanuel Salazar-Cavazos, Patrick O. Byrne, Jason O. Moore, Christopher C. Valley, Kathryn M. Ferguson, Daniel J. Leahy, Diane S. LidkeMark A. Lemmon, 19 October 2017, Cell.
DOI: 10.1016/j.cell.2017.09.017
Never miss a breakthrough: Join the SciTechDaily newsletter.
Follow us on Google and Google News.