Last month, three MIT materials scientists and their colleagues published a paper describing a new artificial intelligence system that can pore through scientific papers and extract “recipes” for producing particular types of materials.
That work was envisioned as the first step toward a system that can originate recipes for materials that have been described only theoretically. Now, in a paper in the journal npj Computational Materials, the same three materials scientists, with a colleague in MIT’s Department of Electrical Engineering and Computer Science (EECS), take a further step in that direction, with a new artificial-intelligence system that can recognize higher-level patterns that are consistent across recipes.
For instance, the new system was able to identify correlations between “precursor” chemicals used in materials recipes and the crystal structures of the resulting products. The same correlations, it turned out, had been documented in the literature.
The system also relies on statistical methods that provide a natural mechanism for generating original recipes. In the paper, the researchers use this mechanism to suggest alternative recipes for known materials, and the suggestions accord well with real recipes.
The first author on the new paper is Edward Kim, a graduate student in materials science and engineering. The senior author is his advisor, Elsa Olivetti, the Atlantic Richfield Assistant Professor of Energy Studies in the Department of Materials Science and Engineering (DMSE). They’re joined by Kevin Huang, a postdoc in DMSE, and by Stefanie Jegelka, the X-Window Consortium Career Development Assistant Professor in EECS.
Sparse and scarce
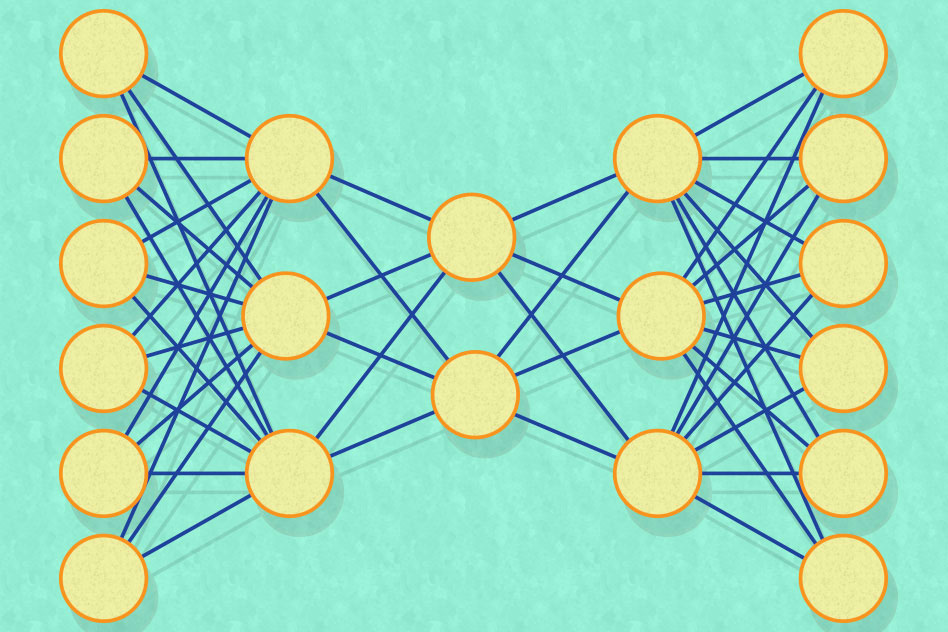
Like many of the best-performing artificial-intelligence systems of the past 10 years, the MIT researchers’ new system is a so-called neural network, which learns to perform computational tasks by analyzing huge sets of training data. Traditionally, attempts to use neural networks to generate materials recipes have run up against two problems, which the researchers describe as sparsity and scarcity.
Any recipe for a material can be represented as a vector, which is essentially a long string of numbers. Each number represents a feature of the recipe, such as the concentration of a particular chemical, the solvent in which it’s dissolved, or the temperature at which a reaction takes place.
Since any given recipe will use only a few of the many chemicals and solvents described in the literature, most of those numbers will be zero. That’s what the researchers mean by “sparse.”
Similarly, to learn how modifying reaction parameters — such as chemical concentrations and temperatures — can affect final products, a system would ideally be trained on a huge number of examples in which those parameters are varied. But for some materials — particularly newer ones — the literature may contain only a few recipes. That’s scarcity.
“People think that with machine learning, you need a lot of data, and if it’s sparse, you need more data,” Kim says. “When you’re trying to focus on a very specific system, where you’re forced to use high-dimensional data but you don’t have a lot of it, can you still use these neural machine-learning techniques?”
Neural networks are typically arranged into layers, each consisting of thousands of simple processing units, or nodes. Each node is connected to several nodes in the layers above and below. Data is fed into the bottom layer, which manipulates it and passes it to the next layer, which manipulates it and passes it to the next, and so on. During training, the connections between nodes are constantly readjusted until the output of the final layer consistently approximates the result of some computation.
The problem with sparse, high-dimensional data is that for any given training example, most nodes in the bottom layer receive no data. It would take a prohibitively large training set to ensure that the network as a whole sees enough data to learn to make reliable generalizations.
Artificial bottleneck
The purpose of the MIT researchers’ network is to distill input vectors into much smaller vectors, all of whose numbers are meaningful for every input. To that end, the network has a middle layer with just a few nodes in it — only two, in some experiments.
The goal of training is simply to configure the network so that its output is as close as possible to its input. If training is successful, then the handful of nodes in the middle layer must somehow represent most of the information contained in the input vector, but in a much more compressed form. Such systems, in which the output attempts to match the input, are called “autoencoders.”
Autoencoding compensates for sparsity, but to handle scarcity, the researchers trained their network on not only recipes for producing particular materials, but also on recipes for producing very similar materials. They used three measures of similarity, one of which seeks to minimize the number of differences between materials — substituting, say, just one atom for another — while preserving crystal structure.
During training, the weight that the network gives example recipes varies according to their similarity scores.
Playing the odds
In fact, the researchers’ network is not just an autoencoder, but what’s called a variational autoencoder. That means that during training, the network is evaluated not only on how well its outputs match its inputs, but also on how well the values taken on by the middle layer accord with some statistical model — say, the familiar bell curve, or normal distribution. That is, across the whole training set, the values taken on by the middle layer should cluster around a central value and then taper off at a regular rate in all directions.
After training a variational autoencoder with a two-node middle layer on recipes for manganese dioxide and related compounds, the researchers constructed a two-dimensional map depicting the values that the two middle nodes took on for each example in the training set.
Remarkably, training examples that used the same precursor chemicals stuck to the same regions of the map, with sharp boundaries between regions. The same was true of training examples that yielded four of manganese dioxide’s common “polymorphs,” or crystal structures. And combining those two mappings indicated correlations between particular precursors and particular crystal structures.
“We thought it was cool that the regions were continuous,” Olivetti says, “because there’s no reason that that should necessarily be true.”
Variational autoencoding is also what enables the researchers’ system to generate new recipes. Because the values taken on by the middle layer adhere to a probability distribution, picking a value from that distribution at random is likely to yield a plausible recipe.
“This actually touches upon various topics that are currently of great interest in machine learning,” Jegelka says. “Learning with structured objects, allowing interpretability by and interaction with experts, and generating structured complex data — we integrate all of these.”
“‘Synthesizability’ is an example of a concept that is central to materials science yet lacks a good physics-based description,” says Bryce Meredig, founder and chief scientist at Citrine Informatics, a company that brings big-data and artificial-intelligence techniques to bear on materials science research. “As a result, computational screens for new materials have been hamstrung for many years by synthetic inaccessibility of the predicted materials. Olivetti and colleagues have taken a novel, data-driven approach to map materials syntheses and made an important contribution toward enabling us to computationally identify materials that not only have exciting properties but also can be made practically in the laboratory.”
The research was supported by the National Science Foundation, the Natural Sciences and Engineering Research Council of Canada, the U.S. Office of Naval Research, the MIT Energy Initiative, and the U.S. Department of Energy’s Basic Energy Science Program.
Reference: “Virtual screening of inorganic materials synthesis parameters with deep learning” by Edward Kim, Kevin Huang, Stefanie Jegelka and Elsa Olivetti, 1 December 2017, npj Computational Materials.
DOI: 10.1038/s41524-017-0055-6
Never miss a breakthrough: Join the SciTechDaily newsletter.
Follow us on Google and Google News.