A large-scale experiment has uncovered the fundamental rules that govern protein stability, opening the door to more rapid development of drugs and enzymes.
Proteins are essential molecular machines that power countless processes in living organisms. They help turn sunlight into energy, support the immune system by fighting off viruses, and much more. Each protein is made from a chain of amino acids, selected from a set of 20 different types. Even a relatively short protein consisting of just 60 amino acids could theoretically be assembled in 10⁷⁸ different ways—a number so vast it rivals the total number of atoms in the known universe (called a quinquavigintillion).
This staggering level of complexity raises a fundamental question: how did evolution manage to select the rare combinations of amino acids that produce proteins that not only fold correctly but also remain stable and perform essential functions? Scientists are also exploring whether these evolutionary rules can be uncovered to help modern researchers design more effective medicines and environmentally friendly catalysts. A new study published in Science offers promising insights into both challenges.
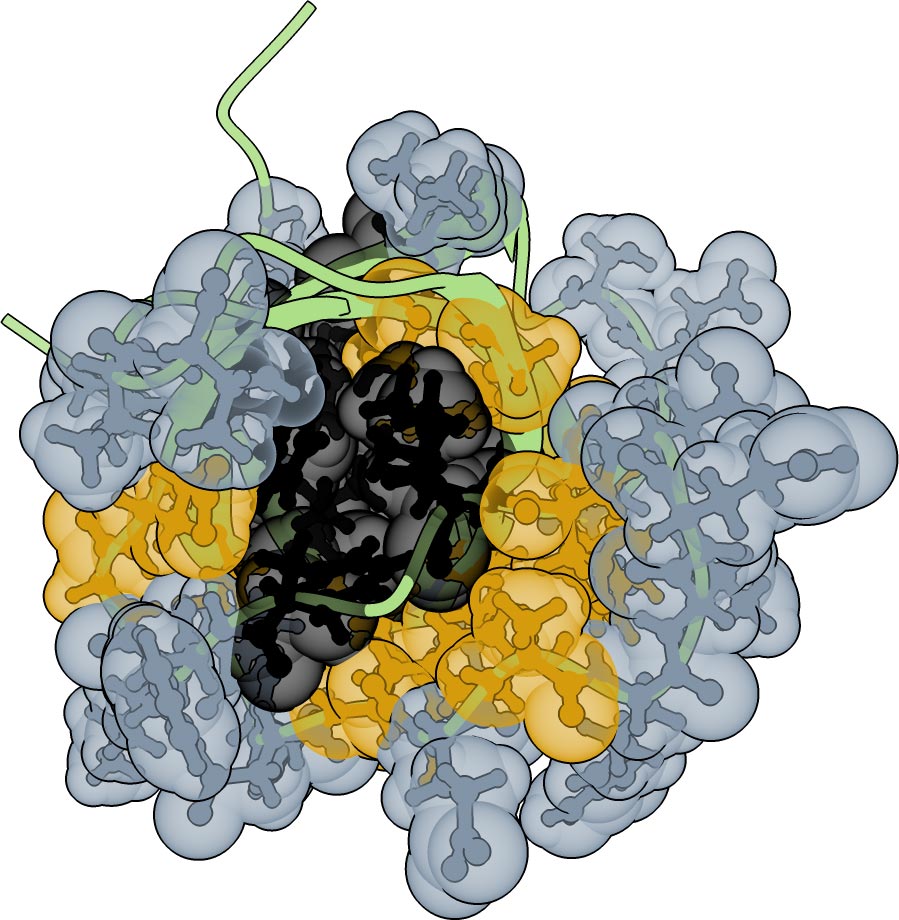
Inside every protein is a tightly packed core that supports its three-dimensional shape. The outer surface, on the other hand, is responsible for most interactions with other molecules. For years, scientists believed that changing any part of the core was like tampering with a building’s support beam. A single mistake could collapse the entire structure. Because the core’s amino acids are closely packed, it seemed reasonable to assume that even small changes could disrupt neighboring regions, triggering a chain reaction of structural failure throughout the protein.
With this classical picture of protein stability, most changes to the building blocks of a protein would set off hidden booby traps and threaten to knock the entire structure out of shape. Given the sheer number of combinations possible, the odds of evolution stumbling onto a safe route to create new proteins seems very small.
A Bold Experiment with the SH3 Domain
The study turns this idea on its head. Researchers at the Centre for Genomic Regulation (CRG) in Barcelona and the Wellcome Sanger Institute in Hinxton, UK, studied a human protein domain (the functional bit of a protein) called FYN-SH3, making hundreds of thousands of variants and testing which ones still folded and worked.
The experiments revealed that SH3 retained its shape and function across thousands of different core and surface combinations. Only a few true, load-bearing amino acids existed in the protein’s core.
“Our data challenges the dogma of proteins being a delicate house of cards. The physical rules governing their stability is more like Lego than Jenga, where a change to one brick threatening to bring the entire structure down is a rare, and crucially, predictable phenomenon,” explains Dr. Albert Escobedo, first author of the study and postdoctoral researcher at the Centre for Genomic Regulation.
The team used the large amount of data generated by their experiments to test whether learning the rules from one protein could help explain the evolution of all related proteins that exist in Nature. They fed the data into a machine-learning algorithm, which helped them create a tool that can predict whether an SH3 sequence will stay stable.
SH3 domains have been diversifying since early multicellular life, roughly one billion years ago. The researchers compared their model against 51,159 natural SH3 sequences found in public databases spanning the entire tree of life, including bacteria, plants, insects, and humans. The algorithm correctly flagged almost all SH3 domains as stable, even when a test sequence shared less than a quarter of the sequence with the human version.
“Evolution didn’t have to sift through an entire universe of sequences. Instead, the biochemical laws of folding create a vast, forgiving landscape for natural selection,” says Dr. Escobedo.
Implications for protein engineering
The field of protein engineering currently relies on companies screening thousands of protein variants with minimal changes, inching forward a few changes at a time, and making the design of new enzymes, drugs, and vaccines slow and expensive.
The confirmation that protein stability follows simpler rules than previously thought can slash the trial-and-error phase for protein design, saving significant time and effort for developing proteins with medical or industrial applications, such as greener catalysts or longer-lasting medicines.
For example, therapeutic enzymes often fail because their surfaces trigger immune flare-ups. Resurfacing these proteins is labor-intensive, requiring lots of trial and error to avoid the scaffold from collapsing and sabotaging a promising design. Now, protein engineers can propose bolder designs, including dozens of simultaneous changes, on computers and walk into the lab already knowing which variants are most likely to survive both folding and functional tests.
“The ability to predict and model protein evolution opens the door to designing biology at industrial speed, challenging the conservative pacing of protein engineering,” explains ICREA Research Professor Ben Lehner, corresponding author of the study with dual affiliation at the Centre for Genomic Regulation (CRG) and the Wellcome Sanger Institute.
Reference: “Genetics, energetics, and allostery in proteins with randomized cores and surfaces” by Albert Escobedo, Gesa Voigt, Andre J. Faure and Ben Lehner, 24 July 2025, Science.
DOI: 10.1126/science.adq3948
Never miss a breakthrough: Join the SciTechDaily newsletter.
Follow us on Google and Google News.