Research identifies impaired muscle regeneration as a key factor in fibrodysplasia ossificans progressiva, driven by a mutation in the ACVR1 gene. This insight shifts potential treatments towards improving muscle repair and reducing unwanted bone growth.
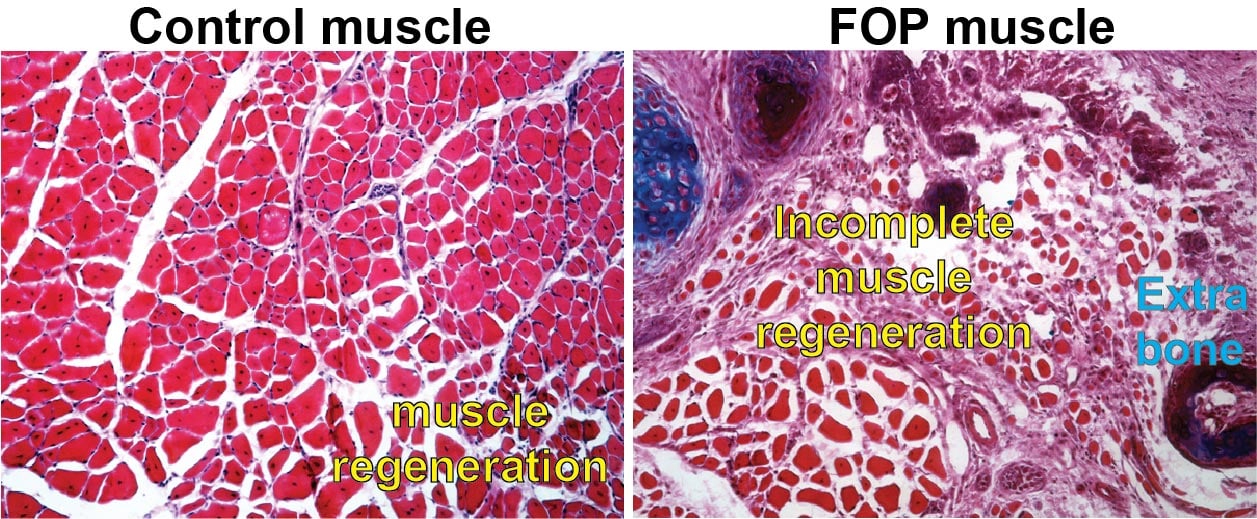
Fibrodysplasia ossificans progressiva (FOP) is a rare disease characterized by extensive bone growth outside of the normal skeleton that pre-empts the body’s normal responses to even minor injuries. It results in what some term a “second skeleton,” which locks up joint movement and could make it hard to breathe. However, new research in mice by a team at the Perelman School of Medicine at the University of Pennsylvania shows that forming extra-skeletal bone might not be the only driver of the disease. Impaired and inefficient muscle tissue regeneration appears to open the door for unwanted bone to form in areas where new muscle should occur after injuries. This discovery opens up the possibility of pursuing new therapies for FOP and was published today in NPJ Regenerative Medicine.
New Insights From Recent Research
“While we have made great strides toward better understanding this disease, this work shows how basic biology can provide great insights into appropriate regenerative medicine therapies,” said the study’s lead author, Foteini Mourkioti, PhD, an assistant professor of Orthopaedic Surgery and Cell and Developmental Biology, as well as the co-director of the Penn Institute for Regenerative Medicine, Musculoskeletal Program.
“From the lab, we’re now able to show that there is potential for a whole new realm of therapies for patients with this devastating condition.”
Genetic Mutations and Cellular Misdirection
About 15 years ago, researchers at Penn – including this study’s co-author, Eileen Shore, PhD, a professor in Orthopaedic Surgery and Genetics and the co-director of the Center for Research in FOP and Related Disorders – discovered that a mutation in the ACVR1 gene was responsible for FOP. In that study, the team found that the mutation changed cells within muscles and connective tissues, misdirecting cells within the tissue to behave like bone cells, resulting in new and unnecessary extra-skeletal bone within the body.
“However, while investigations of how the FOP mutation alters the regulation of cell fate decisions have been extensively pursued in recent years, little attention has been paid to the effects of the genetic mutation on muscle and its impact on the cells that repair muscle injuries,” Shore said. “We were convinced that pursuing research in this area could provide clues not only for preventing extra bone formation but also for improving muscle function and regeneration, bringing new clarity to FOP as a whole.”
Rethinking Muscle Regeneration in FOP
The researchers studied muscle from mice with the same mutation in the ACVR1 gene that people with FOP have. They focused on two specific types of muscle tissue stem cells: fibro-adipogenetic progenitors (FAPs) and muscle stem cells (MuSCs). Typically, muscle injury repair requires a careful balance of these two cell types. Injured tissue responds by an expansion of FAP cells, which are assigned to recruit muscle stem cells that will regenerate the damaged muscle tissue. After about three days, FAPs die off, their job is done. At the same time, MuSCs transition toward a more mature, differentiated state, called muscle fiber, essential to the organized movement of our muscles.
In the mice with the ACVR1 mutation that Mourkioti, Shore, and their co-authors studied, apoptosis – the process through which FAP cells die as a part of proper muscle regeneration – had slowed significantly, leading to a high presence of FAPs past their usual lifespan. This altered their balance with the MuSCs. The injured tissue also showed a diminished capacity for muscle stem cell maturation and, as a result, muscle fibers were considerably smaller in mice carrying the ACVR1 mutation compared to muscle fibers in mice without the mutation.
“The prolonged persistence of diseased FAPs within the regenerating muscle contributes to the altered muscle environment in FOP, which reduces muscle regeneration and allows the over-abundant FAPs to contribute to the formation of extra-skeletal bone,” Mourkioti said. “This provides a completely new perspective on how excess extra-skeletal bone is formed – and how it could be prevented.”
The current targets for treating FOP focus on slowing extra-skeletal bone growth. This research may provide a pivotal new direction. “We propose that therapeutic interventions should consider promoting the regenerating potential of muscles together with the reduction of ectopic bone formation,” Shore and Mourkioti wrote. “By addressing both stem cell populations and their roles in the origin of FOP, there is the possibility of greatly enhanced therapies.”
Reference: “Dynamics of skeletal muscle-resident stem cells during myogenesis in fibrodysplasia ossificans progressive” by Alexandra Stanley, Elisia D. Tichy, Jacob Kocan, Douglas W. Roberts, Eileen M. Shore and Foteini Mourkioti, 14 January 2022, npj Regenerative Medicine.
DOI: 10.1038/s41536-021-00201-8
This study was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01‐AR041916‐15S1, F31-AR069982), the National Institutes of Health (R01‐AR071399, NIH P30-AR069619), and the International Fibrodysplasia Ossificans Progressiva Association (IFOPA).
Other authors in the study include Alexandra Stanley, Elisia Tichy, Jacob Kocan, and Douglas Roberts.
Never miss a breakthrough: Join the SciTechDaily newsletter.
Follow us on Google and Google News.