A newly discovered molecule disrupts cancer cells’ ability to repair DNA by triggering the breakdown of key proteins.
Cancer cells can survive by repairing damage to their DNA—even damage that would normally be fatal. A key defense mechanism is homologous recombination, a highly accurate repair process that fixes broken DNA using proteins such as RAD51 and CHK1. Treatments like PARP inhibitors target this weakness, but many tumors eventually recover their repair ability and become resistant.
A research team led by Kyungjae Myung at the Center for Genomic Integrity within the Institute for Basic Science (IBS), working with Joo-Yong Lee (Chungnam University), has identified a new way to overcome this resistance. Their findings show that cancer cells can be made vulnerable again, not by changing genetic mutations, but by disrupting the stability of the DNA repair machinery itself.
DNA repair proteins are constantly regulated to maintain a balance between fixing damage and preserving genome stability. The researchers found that this balance can be intentionally disturbed.
Discovery of a Key Molecule
Using a cell-based screening method to study replication stress responses, the team identified a small molecule called UNI418. When applied to cancer cells, UNI418 sharply reduced levels of key repair proteins, including RAD51 and CHK1. As these proteins declined, the cells lost their ability to repair DNA efficiently.
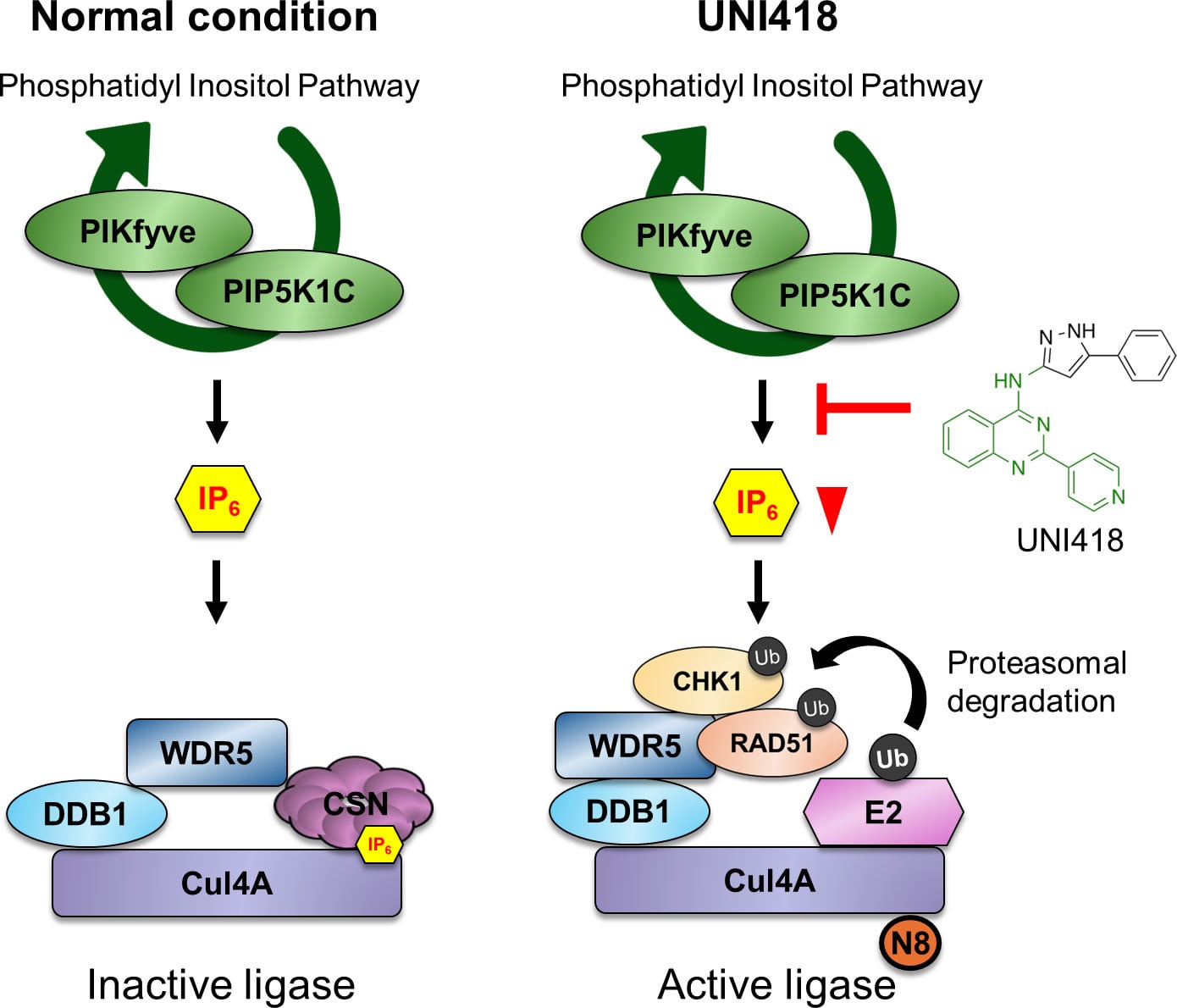
Further investigation showed that UNI418 activates a protein degradation system known as the Cul4A ubiquitin ligase complex, which marks specific proteins for destruction. This process effectively breaks down the DNA repair system from within.
Co-corresponding author Professor Joo-Yong Lee stated, “We identified a mechanism in which key DNA repair proteins are actively degraded inside the cell. This provides a new way to regulate homologous recombination beyond genetic mutations.”
The Role of Cellular Metabolism
The researchers then explored how this degradation process is triggered. UNI418 interferes with a signaling pathway involved in inositol phosphate metabolism, lowering levels of a molecule called IP6. Under normal conditions, IP6 suppresses Cul4A activity. When IP6 levels drop, this suppression is removed, allowing the degradation system to activate.
Cul4A, along with its adaptor protein WDR5, then targets DNA repair proteins such as RAD51 for destruction, shutting down homologous recombination.
This creates a state similar to DNA repair deficiency, even in cancer cells that had previously regained repair function. This effect is especially relevant for overcoming resistance to PARP inhibitors, a major challenge in cancer treatment.
The team tested whether this strategy could improve therapy outcomes. In several cell-based experiments, UNI418 made cancer cells more sensitive to PARP inhibitors. It also worked in cells that had already developed resistance, restoring their response to treatment.
Co-corresponding author Kyungjae Myung added, “By weakening the DNA repair system, we can re-sensitize tumors that have become resistant to existing therapies. This suggests a new strategy for expanding the effectiveness of PARP inhibitors.”
The findings were also confirmed in animal models. In tumor xenograft experiments, UNI418 reduced tumor growth, especially when combined with the PARP inhibitor Olaparib. This effect was observed even in models designed to reflect treatment-resistant cancers.
Broader Implications for Cancer Therapy
The findings were also confirmed in animal models. In tumor xenograft experiments, UNI418 reduced tumor growth, especially when combined with the PARP inhibitor Olaparib. This effect was observed even in models designed to reflect treatment-resistant cancers.
These results show that cancer cells still depend on DNA repair systems even after becoming resistant, and that disrupting protein stability can expose this dependence.
The study also highlights a previously unknown link between cellular metabolism and DNA repair. By connecting IP6 signaling with the Cul4A-driven degradation pathway, the researchers identified a new mechanism that helps regulate genome stability.
Co-corresponding author Kyungjae Myung remarked, “This study demonstrates that controlling the stability of DNA repair proteins can directly impact cancer cell survival. It also highlights a new therapeutic direction for overcoming drug resistance.”
Overall, the findings suggest that drug-resistant cancers can be made vulnerable again by breaking down the systems they use to repair DNA rather than altering their genes. Although UNI418 will require further development, the mechanism described in this study offers a strong foundation for future combination therapies.
Reference: “Targeting IP6 signaling to destabilize homologous recombination proteins to overcome PARP inhibitor resistance” by Seon-gyeong Lee, Yuri Seo, Seula Jeong, Yuheon Chung, Sukyeong Kong, Minyoung Kim, Joon Ho Rhlee, Sihyeon Um, Bijoy P. Mathew, Saikat Maiti, Malleswara Rao Kuram, Mohamed Ahmed Abozeid, Areum Park, Ji-Na Yoo, Keon Woo Khim, Kyuwon Son, Enkhzul Amarsanaa, Kyunghan Kim, Sehoon Hong, Jiyeon Choi, In Bae Park, Eun A. Lee, Ji Hwan Jeon, Jun Hong Park, Joo Seok Han, Chan Young Park, Seyun Kim, Jang Hyun Choi, Sung You Hong, Min-Duk Seo, Hyuk Lee, Joo-Yong Lee and Kyungjae Myung, 4 April 2026, Nature Communications.
DOI: 10.1038/s41467-026-71421-z
The study was funded by the Institute for Basic Science.
Never miss a breakthrough: Join the SciTechDaily newsletter.
Follow us on Google and Google News.