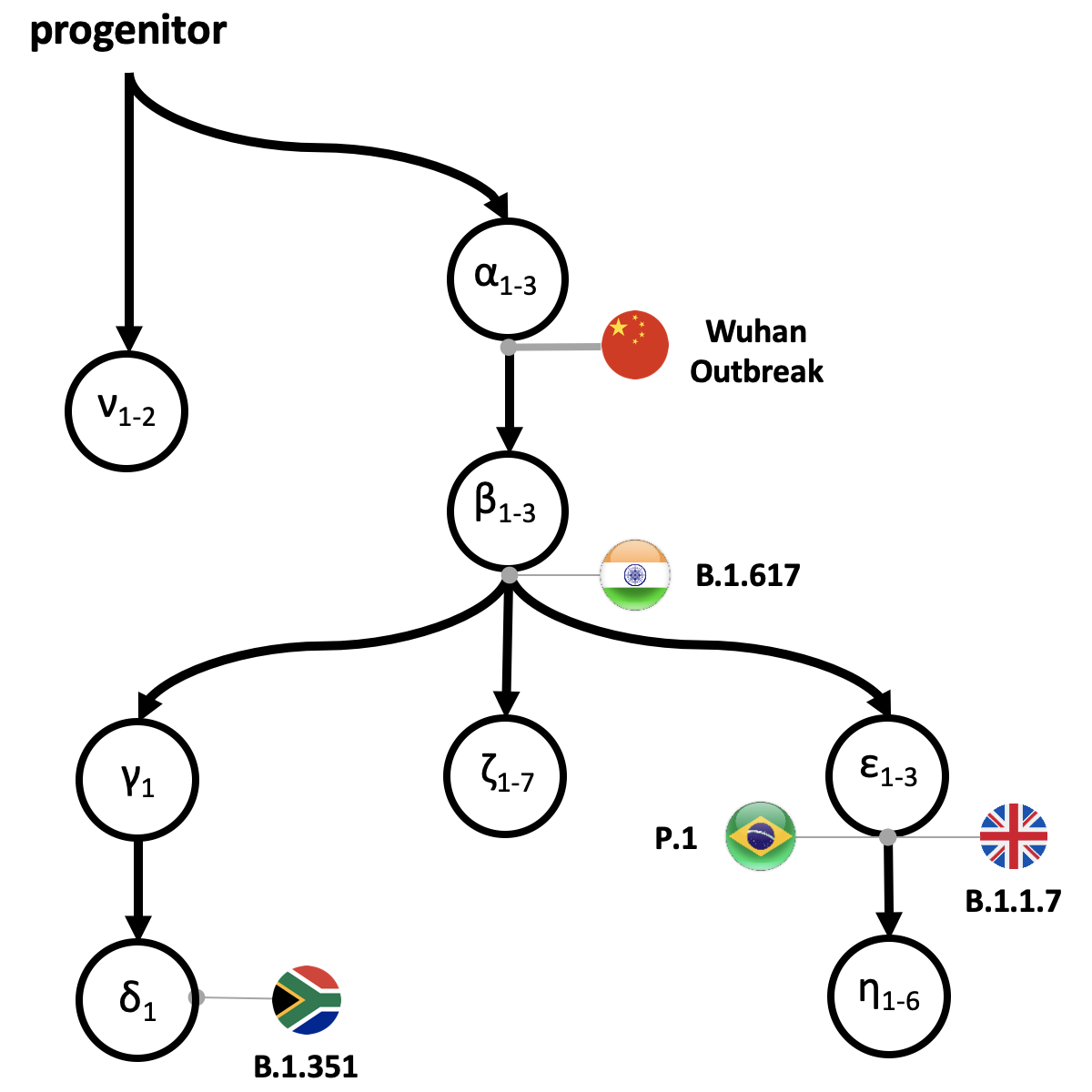
The progenitor (proCoV2) virus and its initial descendants arose in China, based on the earliest mutations of proCoV2 and their locations, which were traced back to occur 6-8 weeks prior to the Wuhan China outbreak. Furthermore, the science team also demonstrated that a population of strains with at least three mutational differences (alpha 1-3) from proCoV2 existed at the time of the first detection of COVID-19 cases in China. The current major variants of interest including the UK (B.1.1.1.7), South African (B.1.351), South American (P.1) and now, Indian (B.1.617) are shown within the pedigree. These variants have not only come to replace prior dominant strains in their respective regions, but still threaten world health due to their potential to escape today’s vaccines and therapeutics. Credit: Sudhir Kumar, Temple University
New study traces back the progenitor genomes causing COVID-19 and geospatial spread.
In the field of molecular epidemiology, the worldwide scientific community has been steadily sleuthing to solve the riddle of the early history of SARS-CoV-2. Despite recent efforts by the World Health Organization, no one to date has identified the first case of human transmission, or “patient zero” in the COVID-19 pandemic.
Finding the earliest possible case is needed to better understand how the virus may have jumped from its animal host first to infect humans as well as the history of how the SARS-CoV-2 viral genome has mutated over time and spread globally.
Since the first SARS-CoV-2 virus infection was detected in December 2019, well over a million genomes of SARS-CoV-2 have been sequenced worldwide, revealing that the coronavirus is mutating, albeit slowly, at a rate of 25 mutations per genome per year. The sheer number of emerging variants, including the UK (B.1.1.1.7), South African (B.1.351), South American (P.1) and now, Indian (B.1.617) have not only come to replace prior dominant strains in their respective regions, but still threaten world health due to their potential to escape today’s vaccines and therapeutics.
“The SARS-CoV-2 virus has already infected more than 145 million people and caused 3 million deaths across the world,” said Sudhir Kumar, director of the Institute for Genomics and Evolutionary Medicine, Temple University. “We set out to find the genetic common ancestor of all these infections, which we call the progenitor genome.”
This progenitor genome (proCoV2) is the mother of all SARS-CoV-2 coronaviruses that has infected and continues to infect people today.
In the absence of patient zero, Kumar and his research team now may have found the next best thing to aid the worldwide molecular epidemiology detective work. “We reconstructed the genome of the progenitor and its early pedigree by using a big dataset of coronavirus genomes obtained from infected individuals since December 2019,” said Kumar, the lead author of a new study, appearing in advanced online edition of the journal Molecular Biology and Evolution.
They found that the progenitor gave rise to a family of coronavirus strains, whose members included the strains found in Wuhan, China, in December 2019. “In essence, the events in December in Wuhan, China, represented the first superspreader event of a virus that had all the tools necessary to cause a worldwide pandemic right out of the box,” said Kumar.
Kumar’s group estimates that the SARS-CoV-2 progenitor was already circulating with an earlier timeline — at least 6 to 8 weeks prior to the first genome sequenced in China, known as Wuhan-1. “This timeline puts the presence of proCoV2 in late October 2019, which is consistent with the report of a fragment of spike protein identical to Wuhan-1 in early December in Italy, among other evidence,” said Sayaka Miura, a senior author of the study.
“We have found progenitor genetic fingerprint in January 2020 and later in multiple coronavirus infections in China and the USA. The progenitor was spreading worldwide months before and after the first reported cases of COVID-19 in China,” said Pond.
Besides their findings on SARS-CoV-2’s early history, Kumar’s group also has developed intuitive mutational fingerprints and Greek symbol classification (ν, α, β, γ, δ, and ε) to simplify the categorization of the major strains, sub-strains and variants infecting an individual or colonizing a global region. This may help scientists better trace and provide context for the order of emergence of new variants.
“Overall, our mutational fingerprinting and nomenclature provide a simple way to glean the ancestry of new variants as compared to phylogenetic designations, e.g., B.1.351 and B.1.1.7,” said Kumar.
For example, an α fingerprint refers to genomes that one or more of the α variants and no other subsequent major variants, and αβ fingerprint refers to genomes that contain all α, at least one β variant, and no other major variants.
“With our tools, we observed the spread and replacement of prevailing strains in Europe (αβε with αβζ) and Asia (α with αβε), the preponderance of the same strain for most of the pandemic in North America (αβ-δ), and the continued presence of multiple high-frequency strains in Asia and North America,” said Pond.
Getting to the root of the problem
To identify the progenitor genome, they used an approach not applied to SARS-CoV-2 previously, called mutation order analysis. The technique, which is used extensively in cancer research, relies on a clonal analysis of mutant strains and the frequency in which pairs of mutations appear together to find the root of the virus.
Many previous attempts in analyzing such large datasets were not successful because of “the focus on building an evolutionary tree of SARS-CoV-2,” says Kumar. “This coronavirus evolves too slow, the number of genomes to analyze is too large, and the data quality of genomes is highly variable. I immediately saw parallels between the properties of these genetic data from coronavirus with the genetic data from the clonal spread of another nefarious disease, cancer.”
Kumar and Miura have developed and investigated many techniques for analyzing genetic data from tumors in cancer patients. They adapted and innovated these techniques to build a trail of mutations that traced back to the progenitor genetic fingerprint. “The mutation tracking approach produced the progenitor and the family history of its major mutation. It is a great example of how big data coupled with biologically informed data mining reveals important patterns,” said Kumar.
An earlier timeline emerges “This progenitor genome had a sequence very different from what some folks are calling the reference sequence, which is what was observed first in China and deposited into the GISAID SARS-CoV-2 database,” said Kumar.
The closest match was to eight genomes sampled 26 to 80 days after the earliest sampled virus from 24 December 2019. Multiple close matches were found in all sampled continents and detected as late as June 2020 (pandemic day 181) in South America. Overall, 140 genomes Kumar’s group analyzed all contained only synonymous differences from proCoV2. That is, all their proteins were identical to the corresponding proCoV2 proteins in the amino acid sequence. A majority (93 genomes) of these protein-level matches were from coronaviruses sampled in China and other Asian countries.
These spatiotemporal patterns suggested that proCoV2 already possessed the full repertoire of protein sequences needed to infect, spread, and persist in the global human population.
They found the proCoV2 virus and its initial descendants arose in China, based on the earliest mutations of proCoV2 and their locations. Furthermore, they also demonstrated that a population of strains with at least three mutational differences from proCoV2 existed at the time of the first detection of COVID-19 cases in China. With estimates of SARS-CoV-2 acquiring 25 mutations per year, this meant that the virus must already have been infecting people several weeks before the December 2019 cases.
Mutational signatures
Because there was strong evidence of many mutations before the ones found in the reference genome, Kumar’s group had to come up with a new nomenclature of mutational signatures to classify SARS-CoV-2 and account for these by introducing a series of Greek letter symbols to represent each one.
For example, they found that the emergence of α SARS-CoV-2 genome variants came before the first reports of COVID-19. This strongly implies the existence of some sequence diversity in the ancestral SARS-CoV-2 populations. All 17 of the genomes sampled from China in December 2019, including the designated SARS-CoV-2 reference genome, carry all three α variants. But, 1,756 genomes without α variants were sampled across the world until July 2020. Therefore, the earliest sampled genomes (including the designated reference) were not the progenitor strains.
It also predicts the progenitor genome had offspring that were spreading worldwide during the earliest phases of COVID-19. It was ready to infect right from the start.
“The progenitor had all the ability it needed to spread,” said Pond. “There is an overabundance of non-synonymous changes in the population. What happened between bats and humans remains unclear, but proCoV2 could already infect at pandemic scales.”
A global spread
Altogether, they have identified seven major evolutionary lineages and the episodic nature of their global spread. The proCoV2 genome gave rise to many major offspring lineages, some of which arose in Europe and North America after the likely genesis of the ancestral lineages in China.
“Asian strains founded the whole pandemic,” said Kumar. “But over time, many variants that evolved elsewhere are now infecting Asia much more.”
Their mutational-based analyses also established that North American coronaviruses harbor very different genome signatures than those prevalent in Europe and Asia.
“This is a dynamic process,” said Kumar. “Clearly, there are very different pictures of spread that are painted by the emergence of new mutations, the three εs, γ&delta, which we found to occur after the spike protein change (a β mutation). Scientists are still figuring out if any functional properties of these mutations have sped up the pandemic.”
Remarkably, the mutational signature of αβ-δ has remained the dominant lineage in North America since April 2020, in contrast to the turnover seen in Europe and Asia. More recently, novel fast-spreading variants including an S protein variant (N501Y) from South Africa and the UK (B.1.1.17) have rapidly increased. Coronaviruses with N501Y variant in South Africa carry the αβγδ genetic fingerprint, whereas those in the UK carry the αβε genetic fingerprint, according to their classification scheme. “Therefore, αβ ancestor continues to give rise to many major offshoots of this coronavirus.” Said Kumar.
Real-time updates
The MBE study relied on three snapshots were retrieved from GISAID on July 7, 2020, (a dataset of 60,332 genomes), October 12, 2020, (contained 133,741 genomes), and finally, an expanded dataset of 172,480 genomes sampled on December 30, 2020.
Moving forward, they will continue to refine their results as new data becomes available.
“More than a million SARS-CoV-2 genomes are sequenced now,” said Pond. “The power of this approach is that the more data you have, the more easily you can tell the precise frequency of individual mutations and mutation pairs. These variants that are produced, the single nucleotide variants, or SNVs, their frequency, and history can be told very well with more data. Therefore, our analyses infer a credible root for the SARS-CoV-2 phylogeny.”
The MBE study is part of their effort to maintain a continuous, live real-time monitoring of SARS-CoV-2 genomes, which has now grown to include more than 350,000 genomes.
“We have set up a live dashboard showing regularly updated results because the processes of data analysis, manuscript preparation, and peer-review of scientific articles are much slower than the pace of expansion of SARS-CoV-2 genome collection,” said Pond. “We also provide a simple “in-the-browser” tool to classify any SARS-CoV-2 genome based on key mutations derived by the MOA analysis.
“These findings and our intuitive mutational fingerprints and barcodes of SARS-CoV-2 strains have overcome daunting challenges to develop a retrospective on how, when and why COVID-19 has emerged and spread, which is a prerequisite to creating remedies to overcome this pandemic through the efforts of science, technology, public policy and medicine,” said Kumar.
Reference: “An evolutionary portrait of the progenitor SARS-CoV-2 and its dominant offshoots in COVID-19 pandemic” by Sudhir Kumar, Qiqing Tao, Steven Weaver, Maxwell Sanderford, Marcos A Caraballo-Ortiz, Sudip Sharma, Sergei L K Pond and Sayaka Miura, 4 May 2021, Molecular Biology and Evolution.
DOI: 10.1093/molbev/msab118







So, they did statistical computations to contradict scientific observation and common sense.
All early last year just one strain that rapidly took over China was worldwide. Variants didn’t appear until late last year, and each had its own initial region where it was found and became common.
@Dora
Variants become popular late last year but were very much present before that.
You can follow IHU infectious disease center infos that were sequencing genomes since pretty much the beginning and were at their 4th dominant strain back in september.