
Despite advances in sequencing technologies and computational methods in the past decade, researchers have uncovered genomes for just a small fraction of Earth’s microbial diversity. Because most microbes cannot be cultivated under laboratory conditions, their genomes can’t be sequenced using traditional approaches. Identifying and characterizing the planet’s microbial diversity is key to understanding the roles of microorganisms in regulating nutrient cycles, as well as gaining insights into potential applications they may have in a wide range of research fields.
A public repository of 52,515 microbial draft genomes generated from environmental samples around the world, expanding the known diversity of bacteria and archaea by 44%, is now available and described on November 9, 2020, in Nature Biotechnology. Known as the GEM (Genomes from Earth’s Microbiomes) catalog, this work results from a collaboration involving more than 200 scientists, researchers at the U.S. Department of Energy (DOE) Joint Genome Institute (JGI), a DOE Office of Science User Facility located at Lawrence Berkeley National Laboratory (Berkeley Lab), and the DOE Systems Biology Knowledgebase (KBase).
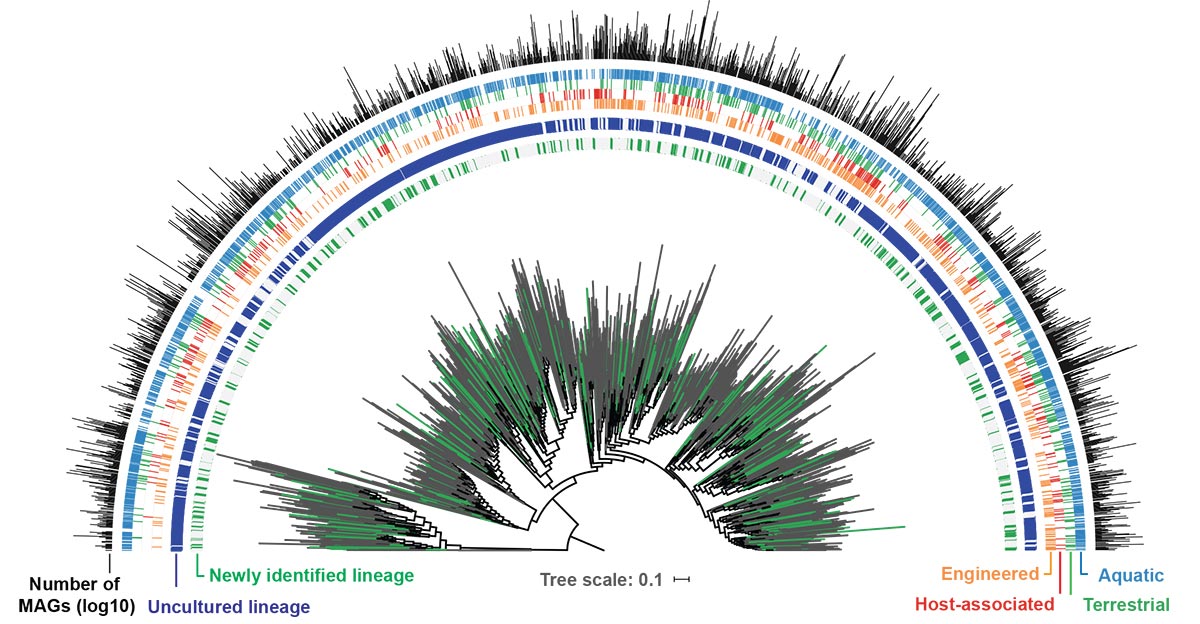
Metagenomics is the study of microbial communities in environmental samples without needing to isolate individual organisms, using various methods for processing, sequencing, and analysis. “Using a technique called metagenome binning, we were able to reconstruct thousands of metagenome-assembled genomes (MAGs) directly from sequenced environmental samples without needing to cultivate the microbes in the lab,” noted Stephen Nayfach, the study’s first author and research scientist in Nikos Kyrpides’ Microbiome Data Science group. “What makes this study really stand out from previous efforts is the remarkable environmental diversity of the samples we analyzed.”
Emiley Eloe-Fadrosh, head of the JGI Metagenome Program and senior author on the study elaborated on Nayfach’s comments. “This study was designed to encompass the broadest and most diverse range of samples and environments, including natural and agricultural soils, human- and animal-host associated, and ocean and other aquatic environments – that’s pretty remarkable.”
Adding Value Beyond Genome Sequences
Much of the data had been generated from environmental samples sequenced by the JGI through the Community Science Program and was already available on the JGI’s Integrated Microbial Genomes & Microbiomes (IMG/M) platform. Eloe-Fadrosh noted that it was a nice example of “big-data” mining to gain a deeper understanding of the data and enhance its value by making data publicly available.
To acknowledge the efforts of the investigators who had done the sampling, Eloe-Fadrosh reached out to more than 200 researchers around the world in accordance with the JGI data use policy. “I felt it is important to acknowledge the significant efforts to collect and extract DNA from these samples, many of which come from unique, difficult-to-access environments, and invited these researchers to be co-authors as part of IMG data consortium,” she said.
Using this massive dataset, Nayfach clustered the MAGs into 18,000 candidate species groups, 70% of which were novel compared to over 500,000 existing genomes available at that time. “Looking across the tree of life, it’s striking how many uncultivated lineages are only represented by MAGs,” he said. “While these draft genomes are imperfect, they can still reveal a lot about the biology and diversity of uncultured microbes.”
Teams of researchers worked on multiple analyses harnessing the genome repository, and the IMG/M team developed several updates and features to mine the GEM catalog. (Watch this IMG webinar on Metagenome Bins to learn more.) One group mined the dataset for novel secondary metabolites of secondary metabolite biosynthetic gene clusters (BGCs), increasing these BGCs in IMG/ABC (Atlas of Biosynthetic Gene Clusters) by 31%. (Listen to this JGI Natural Prodcast episode on genome mining.) Nayfach also worked with another team on predicting host-virus connections between all viruses in IMG/VR (Virus) and the GEM catalog, associating 81,000 viruses – 70% of which had not already been associated with a host – with 23,000 MAGs.
Modeling A New Path for Metagenomics Researchers
Building upon these resources, KBase, a multi-institutional collaborative knowledge creation and discovery environment designed for biologists and bioinformaticians, developed metabolic models for thousands of MAGs. The models are now available in a public Narrative, which provides shareable, reproducible workflows. “Metabolic modeling is a routine analysis for isolate genomes, but has not been done at scale for uncultivated microbes,” said Eloe-Fadrosh, “and we felt that the collaboration with KBase would add value beyond clustering and analysis of these MAGs.

“Just bringing this dataset into KBase has immediate value because people can find the high-quality MAGs and use them to inform future analyses,” said José P. Faria, a KBase computational biologist at Argonne National Laboratory. “The process of building a metabolic model is simple: you just select a genome or MAG and press a button to build a model from our database of mappings between biochemical reactions and annotations. We look at what was annotated in the genome and at the resulting model to assess the metabolic capabilities of the organism.” (Watch this KBase webinar on metabolic modeling.)
KBase User Engagement lead Elisha Wood-Charlson added that by demonstrating the ease with which metabolic models were generated from the GEM dataset, metagenomics researchers might consider branching into this space. “Most metagenomics researchers might not be willing to dive into an entirely new research field [metabolic modeling], but they might be interested in how biochemistry impacts what they work on. The genomics community can now explore metabolism using KBase’s easy path from genomes or MAGs to modeling that may not have been considered,” she said.
A Community Resource for Facilitating Research
Kostas Konstantinidis of Georgia Institute of Technology, one of the co-authors whose data were part of the catalog, “I don’t think there are many institutions that can do this kind of large-scale metagenomics and that have the capacity for large scale analyses. The beauty of this study is that it’s done at this scale that individual labs cannot do, and it gives us new insights into microbial diversity and function.”

He is already finding ways to utilize the catalog in his own research on how microbes respond to climate change. “With this dataset I can see where every microbe is found, and how abundant it is. That’s very useful for my work and for others doing similar research.” Additionally, he’s interested in expanding the diversity of the reference database he’s developing called the Microbial Genomes Atlas to allow for more robust analyses by adding the MAGs.
“This is a great resource for the community,” Konstantinidis added. “It’s a dataset that is going to facilitate many more studies subsequently. And I hope JGI and other institutions continue to do this kind of projects.”
Reference: “A genomic catalog of Earth’s microbiome” by Stephen Nayfach, Simon Roux, Rekha Seshadri, Daniel Udwary, Neha Varghese, Frederik Schulz, Dongying Wu, David Paez-Espino, I-Min Chen, Marcel Huntemann, Krishna Palaniappan, Joshua Ladau, Supratim Mukherjee, T. B. K. Reddy, Torben Nielsen, Edward Kirton, José P. Faria, Janaka N. Edirisinghe, Christopher S. Henry, Sean P. Jungbluth, Dylan Chivian, Paramvir Dehal, Elisha M. Wood-Charlson, Adam P. Arkin, Susannah G. Tringe, Axel Visel, IMG/M Data Consortium, Tanja Woyke, Nigel J. Mouncey, Natalia N. Ivanova, Nikos C. Kyrpides and Emiley A. Eloe-Fadrosh, 9 November 2020, Nature Biotechnology.
DOI: 10.1038/s41587-020-0718-6
The work also used resources of the National Energy Research Scientific Computing Center (NERSC), another DOE Office of Science User Facility located at Berkeley Lab.
Never miss a breakthrough: Join the SciTechDaily newsletter.
Follow us on Google and Google News.