Results could explain why drugs designed to remove amyloid deposits have failed to stop disease.
According to the National Institute on Aging, Alzheimer’s disease is the most common cause of dementia in older adults and the 7th leading cause of death in the United States. It is a debilitating progressive illness that slowly destroys cognitive function and memory.
The amyloid cascade hypothesis is a leading theory for how Alzheimer’s disease forms. Now, new research findings point in a new direction.
A breakdown in how brain cells rid themselves of waste precedes the buildup of debris-filled plaques known to occur in Alzheimer’s disease, a new study in mice shows.
The field argued for decades that such plaques, containing the protein amyloid beta, built up outside of cells as a crucial first step toward the brain damage observed in Alzheimer’s disease. Led by researchers at NYU Grossman School of Medicine and the Nathan Kline Institute, the new study challenges this idea, known as the amyloid cascade hypothesis.
The latest research findings argue instead that neuronal damage characteristic of Alzheimer’s disease takes root inside cells well before these thread-like amyloid plaques fully form and clump together in the brain.
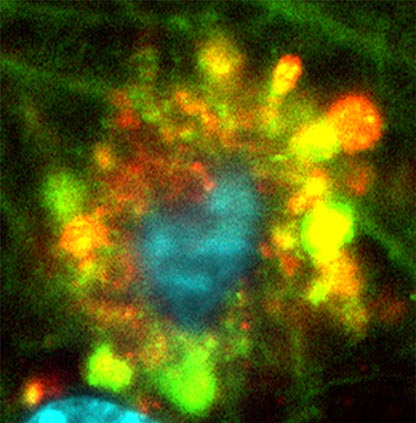
Lysosomes and Cellular Waste Management
Published on June 2, 2022, as the cover article in the journal Nature Neuroscience, the study traced the root dysfunction observed in mice bred to develop Alzheimer’s disease to the brain cells’ lysosomes. These are small sacs inside every cell, filled with acidic enzymes involved in the routine breakdown, removal, and recycling of metabolic waste from everyday cell reactions, as well as from disease. Lysosomes are also key, researchers note, to breaking down and disposing of a cell’s own parts when the cell naturally dies.
As part of the study, scientists tracked decreasing acid activity inside intact mouse cell lysosomes as the cells became injured in the disease. Imaging tests developed at NYU Langone Health and Nathan Kline (to track cellular waste removal) showed that certain brain cell lysosomes became enlarged as they fused with so-called autophagic vacuoles filled with waste that had failed to be broken down. These autophagic vacuoles also contained earlier forms of amyloid beta.
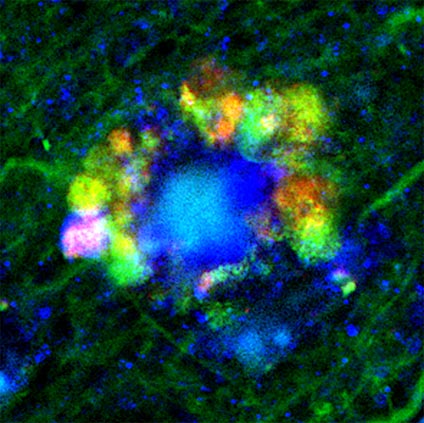
In neurons most heavily damaged and destined for early death as a result, the vacuoles pooled together in “flower-like” patterns, bulging out from the cells’ outer membranes and massing around each cell’s center, or nucleus. Accumulations of amyloid beta formed filaments inside the cell, another hallmark of Alzheimer’s disease. Indeed, researchers observed almost-fully formed plaques inside some damaged neurons.
Challenging the Amyloid Cascade Hypothesis
“Our results for the first time sources neuronal damage observed in Alzheimer’s disease to problems inside brain cells’ lysosomes where amyloid beta first appears,” says study lead investigator Ju-Hyun Lee, PhD.
“Previously, the working hypothesis mostly attributed the damage observed in Alzheimer’s disease to what came after amyloid buildup outside of brain cells, not before and from within neurons,” says Lee, a research assistant professor in the Department of Psychiatry and NYU Langone Health and research scientist at Nathan Kline.

Implications for Alzheimer’s Treatment Strategies
“This new evidence changes our fundamental understanding of how Alzheimer’s disease progresses; it also explains why so many experimental therapies designed to remove amyloid plaques have failed to stop disease progression, because the brain cells are already crippled before the plaques fully form outside the cell,” says study senior investigator Ralph Nixon, MD, PhD.
“Our research suggests that future treatments should focus on reversing the lysosomal dysfunction and rebalancing acid levels inside the brain’s neurons,” says Nixon, a professor in the Department of Psychiatry and the Department of Cell Biology at NYU Langone, as well as director of the Center for Dementia Research at Nathan Kline.
Researchers say they are already working on experimental therapies to treat the lysosomal problems observed in their studies.
A recent study (published in April in Science Advances) by the NYU Langone team sourced one cause of the cell’s waste disposal problems to a gene called PSEN1. The gene has long been known to cause Alzheimer’s disease, but its additional role in causing the illness (through lysosomal dysfunction) is only now becoming clear.
Their recent work also showed that the neuronal damage in a PSEN1 mouse model of Alzheimer’s disease could be reversed by restoring proper acid levels in lysosomes.
This work is covered by United States Patent 9,265,735 that is directed to methods of treating Alzheimer’s disease based on reversing lysosomal de-acidification, the underlying cause of waste buildup. The terms and conditions of the patent are being managed in accordance with the policies of the health system.
According to the National Institute on Aging, more than 6 million Americans, most of them age 65 or older, have dementia, a progressive loss of thinking, remembering, and reasoning, due to Alzheimer’s disease.
Reference: “Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Aβ in neurons, yielding senile plaques” by Ju-Hyun Lee, Dun-Sheng Yang, Chris N. Goulbourne, Eunju Im, Philip Stavrides, Anna Pensalfini, Han Chan, Cedric Bouchet-Marquis, Cynthia Bleiwas, Martin J. Berg, Chunfeng Huo, James Peddy, Monika Pawlik, Efrat Levy, Mala Rao, Mathias Staufenbiel and Ralph A. Nixon, 2 June 2022, Nature Neuroscience.
DOI: 10.1038/s41593-022-01084-8
Funding for these studies was provided by National Institute of Health grants P01AG017617, P50AG025688, and R01AG062376.
Besides Lee and Nixon, other NYU Langone and Nathan Kline study investigators involved in this research are Dun-Sheng Yang, Chris Goulbourne, Eunju Im, Philip Stavrides, Ann Pensalfini, Cynthia Bleiwas, Martin Berg, Chunfeng Huo, James Peddy, Monika Pawlik, Efrat Levy, and Mala Rao. Additional co-investigators are Han Chan and Cedric Bouchet-Marquis, at Thermo-Fisher Scientific in Hillsboro, Ore.; and Mathias Staufenbiel, at the University of Tubingen in Germany.
Never miss a breakthrough: Join the SciTechDaily newsletter.
Follow us on Google and Google News.
1 Comment
How could the Patent Office award a patent so ridiculously broad?