The peptide blocks a hyperactive brain enzyme that contributes to the neurodegeneration seen in Alzheimer’s and other diseases.
MIT neuroscientists have found a way to reverse neurodegeneration and other symptoms of Alzheimer’s disease by interfering with an enzyme that is typically overactive in the brains of Alzheimer’s patients.
When the researchers treated mice with a peptide that blocks the hyperactive version of an enzyme called CDK5, they found dramatic reductions in neurodegeneration and DNA damage in the brain. These mice also showed improvements in their ability to perform tasks such as learning to navigate a water maze.
“We found that the effect of this peptide is just remarkable,” says Li-Huei Tsai, director of MIT’s Picower Institute for Learning and Memory and the senior author of the study. “We saw wonderful effects in terms of reducing neurodegeneration and neuroinflammatory responses, and even rescuing behavior deficits.”
With further testing, the researchers hope that the peptide could eventually be used as a treatment for patients with Alzheimer’s disease and other forms of dementia that have CDK5 overactivation. The peptide does not interfere with CDK1, an essential enzyme that is structurally similar to CDK5, and it is similar in size to other peptide drugs that are used in clinical applications.
Picower Institute Research Scientist Ping-Chieh Pao is the lead author of the paper, which was published on April 12 in the Proceedings of the National Academy of Sciences.
Targeting CDK5
Tsai has been studying CDK5’s role in Alzheimer’s disease and other neurodegenerative diseases since early in her career. As a postdoc, she identified and cloned the CDK5 gene, which encodes a type of enzyme known as a cyclin-dependent kinase. Most of the other cyclin-dependent kinases are involved in controlling cell division, but CDK5 is not. Instead, it plays important roles in the development of the central nervous system, and also helps to regulate synaptic function.
CDK5 is activated by a smaller protein that it interacts with, known as P35. When P35 binds to CDK5, the enzyme’s structure changes, allowing it to phosphorylate — add a phosphate molecule to — its targets. However, in Alzheimer’s and other neurodegenerative diseases, P35 is cleaved into a smaller protein called P25, which can also bind to CDK5 but has a longer half-life than P35.
When bound to P25, CDK5 becomes more active in cells. P25 also allows CDK5 to phosphorylate molecules other than its usual targets, including the Tau protein. Hyperphosphorylated Tau proteins form the neurofibrillary tangles that are one of the characteristic features of Alzheimer’s disease.
In previous work, Tsai’s lab has shown that transgenic mice engineered to express P25 develop severe neurodegeneration. In humans, P25 has been linked to several diseases, including not only Alzheimer’s but also Parkinson’s disease and frontotemporal dementia.
Pharmaceutical companies have tried to target P25 with small-molecule drugs, but these drugs tend to cause side effects because they also interfere with other cyclin-dependent kinases, so none of them have been tested in patients.
The MIT team decided to take a different approach to targeting P25, by using a peptide instead of a small molecule. They designed their peptide with a sequence identical to that of a segment of CDK5 known as the T loop, which is a structure critical to the binding of CDK5 to P25. The entire peptide is only 12 amino acids long — slightly longer than most existing peptide drugs, which are five to 10 amino acids long.
“From a peptide drug point of view, usually smaller is better,” Tsai says. “Our peptide is almost within that ideal molecular size.”
Dramatic Effects
In tests in neurons grown in a lab dish, the researchers found that treatment with the peptide led to a moderate reduction in CDK5 activity. Those tests also showed that the peptide does not inhibit the normal CDK5-P35 complex, nor does it affect other cyclin-dependent kinases.
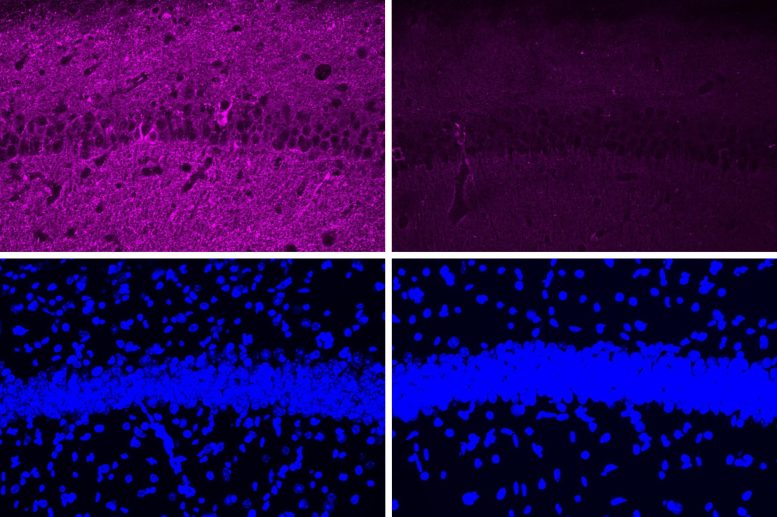
When the researchers tested the peptide in a mouse model of Alzheimer’s disease that has hyperactive CDK5, they saw a myriad of beneficial effects, including reductions in DNA damage, neural inflammation, and neuron loss. These effects were much more pronounced in the mouse studies than in tests in cultured cells.
The peptide treatment also produced dramatic improvements in a different mouse model of Alzheimer’s, which has a mutant form of the Tau protein that leads to neurofibrillary tangles. After treatment, those mice showed reductions in both Tau pathologies and neuron loss. Along with those effects in the brain, the researchers also observed behavioral improvements. Mice treated with the peptide performed much better in a task that required learning to navigate a water maze, which relies on spatial memory, than mice that were treated with a control peptide (a scrambled version of the peptide used to inhibit CDK5-P25).
In those mouse studies, the researchers injected the peptide and found that it was able to cross the blood-brain barrier and reach neurons of the hippocampus and other parts of the brain.
The researchers also analyzed the changes in gene expression that occur in mouse neurons following treatment with the peptide. Among the changes they observed was an increase in expression of about 20 genes that are typically activated by a family of gene regulators called MEF2. Tsai’s lab has previously shown that MEF2 activation of these genes can confer resilience to cognitive impairment in the brains of people with Tau tangles, and she hypothesizes that the peptide treatment may have similar effects.
“Further development of such peptide inhibitors toward a lead therapeutic candidate, if proven to be selective for the target and relatively free of clinical side effects, may eventually lead to novel treatments for neurodegenerative disorders ranging from Alzheimer’s disease to Frontotemporal dementia to Parkinson’s disease,” says Stuart Lipton, a professor of neuroscience at Scripps Research, who was not involved in the study.
Tsai now plans to do further studies in other mouse models of diseases that involve P25-associated neurodegeneration, such as frontotemporal dementia, HIV-induced dementia, and diabetes-linked cognitive impairment.
“It’s very hard to say precisely which disease will most benefit, so I think a lot more work is needed,” she says.
Reference: “A Cdk5-derived peptide inhibits Cdk5/p25 activity and improves neurodegenerative phenotypes” by Ping-Chieh Pao, Jinsoo Seo, Audrey Lee, Oleg Kritskiy, Debasis Patnaik, Jay Penney, Ravikiran M. Raju, Ute Geigenmuller, M. Catarina Silva, Diane E. Lucente, James F. Gusella, Bradford C. Dickerson, Anjanet Loon, Margaret X. Yu, Michael Bula, Melody Yu, Stephen J. Haggarty and Li-Huei Tsai, 12 April 2023, Proceedings of the National Academy of Sciences.
DOI: 10.1073/pnas.2217864120
The research was funded by the National Institutes of Health.
Never miss a breakthrough: Join the SciTechDaily newsletter.
Follow us on Google and Google News.
4 Comments
Fascinating, interesting topics. Makes me want to be involved somehow to be part of it. Many Thanks
Yet another grossly misleading article title, contributing to this site’s lack of credibility in science reporting and more misinformed claims circulating the internet. “Promising experiments on mice” does not equal “cure for Alzheimer’s in humans” as proven by the many thousands of previous promising leads over the past decades. If you can’t word your article titles accurately, don’t publish the article.
I’m confused. Where does the article say “cure for Alzheimer’s in humans.” I also complain about clickbait but this title wasn’t even that bad
“Could eventually” ought to tell you what NONSENSE this is. People with Alzheimer’s are on borrowed time. They’d VOLUNTEER to get rid of its effects, so why are there no human trials announced if it’s so effective? People are dying in the mean time. The problem is (see Saccharin) that mice are not humans and never will be. Not everything that works in a mouse works in a human (see spinal recovery ‘solutions’ that never worked in humans, but work in mice). I don’t know how many articles I’ve seen on “breakthroughs” in everything from Fusion to ageing to cancer and other diseases that NEVER SEEM PAN OUT. One could almost conclude such articles are “click bait”.