
Researchers have identified a versatile enzyme capable of reshaping peptide-based drugs into more stable, longer-lasting forms.
Chemistry researchers at the University of Utah have identified an enzyme called PapB that can “tie off” therapeutic peptides, which are protein-like drugs, into compact ring structures. This process is known as macrocyclization.
According to new research, this approach could help scientists develop stronger and longer-lasting versions of GLP-1 drugs such as semaglutide, the active ingredient in Ozempic and Wegovy, which are used to treat diabetes and obesity.
Forming peptides into rings is especially valuable because these structures improve stability, extend how long drugs remain active in the body, and can enhance how effectively they interact with biological targets. This is according to co-author Karsten Eastman, a research associate in the university’s Department of Chemistry and CEO and co-founder of Sethera Therapeutics.
“Peptides themselves can be extremely difficult to work with because they have a lot of reactive chemical handles. But this is what makes them so great in biology. You can get the type of reaction that you want in the body, but it’s difficult to modify them in hyper-specific ways,” said Eastman, who completed his Ph.D. in 2023 in the lab of Utah chemistry professor Vahe Bandarian. “What we show in the study is an enzymatic method—using a tiny molecular machine to modify or hyper-modify peptides in extremely controlled ways—enabling what we believe will be next-generation peptide therapeutics.”
Eastman and Bandarian, a coauthor of the study, founded Sethera last year to bring their university discoveries to market with support from the National Institutes of Health. Their work was recently recognized by the university’s Technology Licensing Office, which named them the 2025 Founders of the Year for creating the PolyMacrocyclic Peptide (pMCP) Discovery Platform.
A Simpler Alternative to Traditional Methods
Conventional chemical approaches for closing peptide rings are often costly and difficult to apply late in drug development. PapB offers a more straightforward alternative. It forms a specific chemical bond that closes the peptide into a ring without requiring the extra “leader” sequences that many enzymes depend on to identify their targets.
The study, published in ACS Bio & Med Chem Au, explains how the team used PapB, a “radical SAM” (S-adenosyl-L-methionine) enzyme, to link the ends of GLP-1-like peptides through a sulfur-carbon bond called a thioether. Laboratory experiments confirmed that the rings formed successfully, even when the peptides contained nonstandard building blocks commonly used in modern incretin drugs for diabetes treatment.
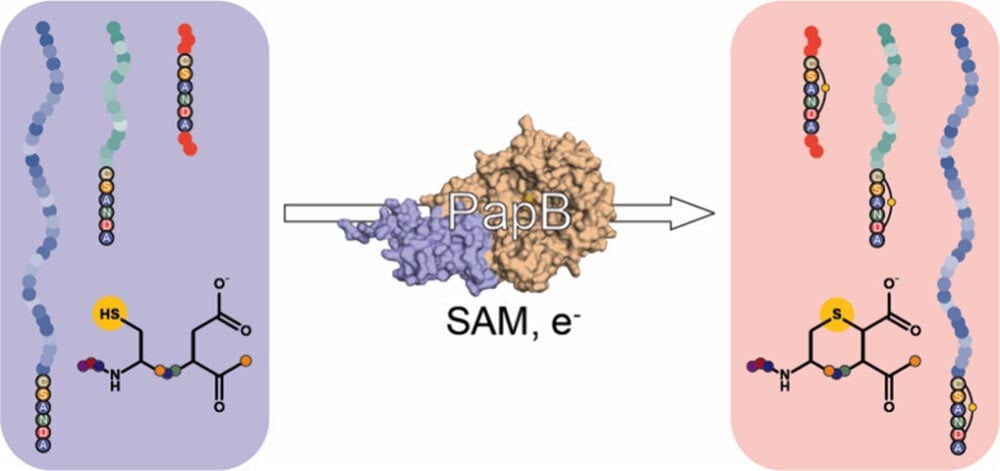
“We were surprised by how flexible the enzyme turned out to be,” said Jake Pedigo, the study’s lead author and a graduate student in the Bandarian lab. “It didn’t need the usual leader sequence, and it still worked even when we swapped in unusual amino acids. That combination of precision and adaptability makes PapB a practical tool for peptide engineering.”
Earlier research from the lab had introduced this peptide-closing approach, and the new work provides clear proof of concept. The researchers tested PapB on three GLP-1-like peptides, and in each case the enzyme converted the open chain into a ring structure. These findings indicate that PapB could act as a flexible biocatalyst for modifying peptides at later stages of drug development.
“The new study ties together a significant amount of research in a new way, enabling an already on-the-market therapeutic to have a specific type of modification that no one has been able to achieve, especially using an enzymatic method,” Eastman said.
This method may also improve peptide stability, which could lead to better therapeutic performance.
Enhancing Drug Longevity in the Body
The human body constantly breaks down proteins using proteases, enzymes that cut peptides into individual amino acids. While this process is essential for normal biology, it can limit how long peptide drugs remain effective.
“You have these peptides that could have a great biological response, but if that biological response only lasts minutes, then all of a sudden you don’t have a good therapeutic,” Eastman said. “By using this enzymatic method to tie off the ends, we are essentially hiding the peptide from some of the most common proteases in the body—which are what breaks down peptides. This would enable the longer half-life.”
Standard chemical methods for forming rings are not always suitable for delicate peptide drugs. Enzymes such as PapB provide a more controlled option, although most were previously believed to require leader sequences to work.
By showing that PapB functions without this requirement, the researchers demonstrate that it could be applied to a broad range of peptides. This flexibility may open the door to new drug designs that are more stable, more targeted, and easier to produce.
“Big pharma’s GLP-1 backbones are already excellent,” Eastman said. “What we’re adding is a clean, late-stage enzymatic step that can make those molecules work even harder. By installing a small, well-defined ring, we can tune how long the drug lasts, how stable it is, and even how it signals—all while staying compatible with the complex structures already in use.”
Reference: “Leader-Independent C-Terminal Modification by a Radical S-Adenosyl-l-methionine Maturase Enables Macrocyclic GLP-1-Like Peptides” by Jacob K. Pedigo, Karsten A. S. Eastman and Vahe Bandarian, 14 October 2025, ACS Bio & Med Chem Au.
DOI: 10.1021/acsbiomedchemau.5c00152
This research was supported by grants from the National Institute of General Medical Sciences.
Never miss a breakthrough: Join the SciTechDaily newsletter.
Follow us on Google and Google News.