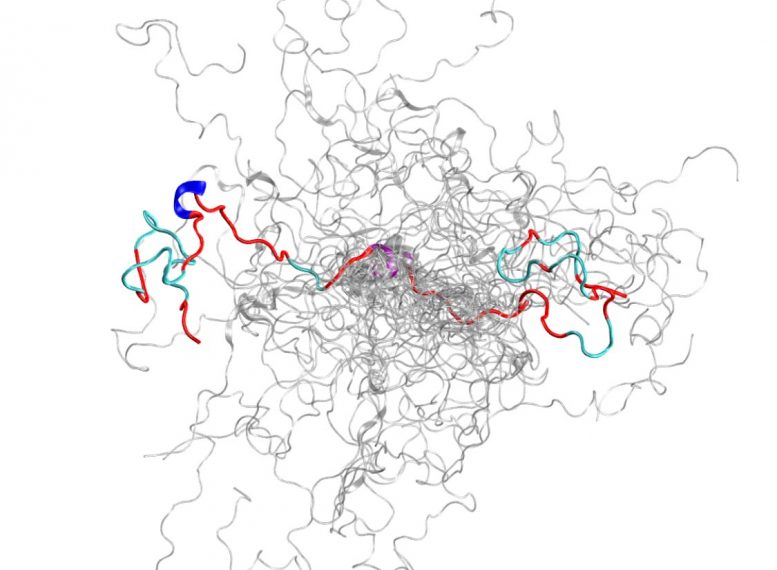
Using the Titan supercomputer and the Spallation Neutron Source at the Department of Energy’s Oak Ridge National Laboratory, scientists have created the most accurate 3D model yet of an intrinsically disordered protein, revealing the ensemble of its atomic-level structures.
As its name indicates, an IDP does not adopt an ordered, static structure like other proteins; instead, it’s flexible and can adopt multiple 3D structures. This lack of a unique structure is necessary for the IDP’s biological function but makes it technically challenging to study. IDPs may be a whole protein or a domain of an otherwise structured protein, and they make up a large portion of human, microbe, and plant proteins.
Loukas Petridis, a staff scientist at the Center for Molecular Biophysics at ORNL, has directed a team of researchers to a new way to create accurate physical models of such flexible biosystems, which can lead to a better understanding of their biological functions. Over the past three years, the team has combined neutron scattering experiments with enhanced sampling molecular dynamics (MD) simulations so computationally demanding that they required the processing power of Titan, the recently decommissioned 27-petaflop Cray XK7 at the Oak Ridge Leadership Computing Facility, a DOE Office of Science User Facility at ORNL.
“To study these IDPs is quite difficult, from both perspectives of experiments and modeling,” said Utsab Shrestha, the lead author of the team’s paper, recently published in the Proceedings of the National Academy of Sciences. “We not only thought about it from experiment or simulation alone, we planned in a way that we would synergize both of these approaches—combine them in a way that we could get more precise information on IDPs. Specifically, simulations helped us to generate an accurate ensemble of IDP at atomic resolution, which is difficult to determine from experiments alone.”
Typically, researchers conduct experiments such as small-angle neutron scattering, small-angle x-ray scattering, or nuclear magnetic resonance to probe flexible biological systems. However, these methods do not provide a detailed atomic-level picture of an IDP’s 3D structures, known as its configurational ensemble. Furthermore, they can only produce ensemble-averaged data, rather than the specific underlying protein structure configurations. Scientists have also performed computer simulations of IDP and compared them with such experiments, hoping to get the same results in order to verify the accuracy of their models.
“But they end up not agreeing with the experiments,” Petridis said. “And because of the discrepancy between the simulations and the experiments, they have to reweight the simulations—they have to adjust the simulation results to make them match the experiments, which is frustrating. That was the state of the art until our work.”
Computer MD simulations conducted by Shrestha used enhanced sampling methods that succeeded in matching not only neutron scattering experiments—conducted by Viswanathan Gurumoorthy and his colleagues at SNS, a DOE Office of Science User Facility at ORNL—but also previously published NMR data. These MD simulations use physics to determine how proteins move. Key to the team’s success was running many MD simulations in parallel on Titan, allowing the simulations to communicate with each other and exchange information.
“This is very important because it allows the simulation to sample a larger configurational space, explore more of the three-dimensional structures in a more efficient way,” Petridis said. “That’s why this enhanced-sampling MD can produce results that the normal MD simulation cannot. We’d have to run a normal MD simulation for years to obtain the same results.”
The IDP that the team chose to study is the N-terminal domain of c-Src kinase, which is a major signaling protein in humans. Mutations in this complex protein have been correlated with cancer, which also makes it an important drug target. While mapping this previously murky domain, the scientists were able to discover new information about its 3D structures that previous methods had not shown. For example, although it is largely disordered, this protein forms transient ordered structures, such as helices.
“The combination of neutron scattering experiments and simulation is very powerful,” Petridis said. “Validation of the simulations by comparison to neutron scattering experiments is essential to have confidence in the simulation results. The validated simulations can then provide detailed information that is not directly obtained by experiments.”
The detailed computer model of the IDP’s 3D structure ensemble opens the door to more experimentation. For example, scientists could simulate the effect of phosphorylation (the addition of a phosphate group to the protein that can regulate the protein’s function) to see what structural changes take place in c-Src kinase that could influence its function. The role of mutations could also be examined: If a researcher changes an amino acid in the chain, how does this affect the structure or the ensemble of structures?
“There are a lot of unanswered questions for c-Src kinase in particular that could be answered in terms of the interactions with other partners—the effect of phosphorylation, the effect of mutations,” Petridis said.
Beyond the potential scientific uses for the model itself, Petridis sees opportunities to apply the use of high-performance computing for running enhanced sampling MD to study the structures of many other important IDPs, which could give insight into their function. And more broadly, the team wants to develop simulation technologies that can reproduce small-angle neutron scattering profiles of even more complex biological systems.
“We don’t want to investigate only the disordered proteins—we want to have much bigger systems that contain ordered and disordered domains that may be interacting with membranes or DNA,” Petridis said. “Neutron scattering is, in my view, the best experimental technique to probe these multi-component systems—for example, a protein that interacts with a membrane or a protein that interacts with DNA. But, still, neutron scattering needs the accurate simulations to better interpret the data.”
###
Coauthors of this study include Utsab R. Shrestha, Puneet Juneja, Qiu Zhang, Viswanathan Gurumoorthy, Jose M. Borreguero, Volker Urban, Xiaolin Cheng, Sai Venkatesh Pingali, Jeremy C. Smith, Hugh M. O’Neill, and Loukas Petridis. Support for this project came from ORNL’s Laboratory Directed Research and Development Program and from DOE’s Office of Science. In addition to using the OLCF’s Titan supercomputer and Spallation Neutron Source, the team performed research at the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility at Lawrence Berkeley National Laboratory.
UT-Battelle LLC manages Oak Ridge National Laboratory for DOE’s Office of Science, the single largest supporter of basic research in the physical sciences in the United States. DOE’s Office of Science is working to address some of the most pressing challenges of our time.
Reference: “Generation of the configurational ensemble of an intrinsically disordered protein from unbiased molecular dynamics simulation” by Utsab R. Shrestha, Puneet Juneja, Qiu Zhang, Viswanathan Gurumoorthy, Jose M. Borreguero, Volker Urban, Xiaolin Cheng, Sai Venkatesh Pingali, Jeremy C. Smith, Hugh M. O’Neill, and Loukas Petridis, 23 September 2019, Proceedings of the National Academy of Sciences.
DOI: 10.1073/pnas.1907251116
Never miss a breakthrough: Join the SciTechDaily newsletter.
Follow us on Google and Google News.